wwPDB 2025 News
Contents
06/25/2025
Extension to EMDB Accession Codes
The EMDB archive is experiencing near-exponential growth. To support this rapid expansion, the wwPDB has been working to prepare our systems for the future and ensure seamless deposition, validation, and dissemination of data in the years ahead.
As part of this effort, we are announcing an important change:
EMDB IDs will be extended to support up to six digits, allowing for identifiers such as EMD-123456. Currently, the archive uses four and five-digit IDs (e.g., EMD-0123, EMD-45678), with support for identifiers up to the EMDB ID: EMD-99999. The change to six-digits will increase our capacity ten-fold and ensure ID availability into the next decade, up to EMD-999999.
We do not anticipate surpassing 99,999 released entries until around 2028, however, we expect to begin assigning six-digit EMDB IDs sometime in 2026. As such, as early as 2026, depositors should anticipate being assigned six-digit IDs within the wwPDB OneDep deposition system, and the user and developer community can expect six-digit EMDB IDs in the EMDB archive.
We appreciate your support as the archive grows and thank you for your contribution to the worldwide effort to archive structural biology data. We remain committed to ensuring a stable, sustainable and scalable infrastructure that supports the continued successes and productivity of the scientific community using structural biology and cryoEM in their research. For questions or feedback, contact the EMDB team at emdbhelp@ebi.ac.uk.
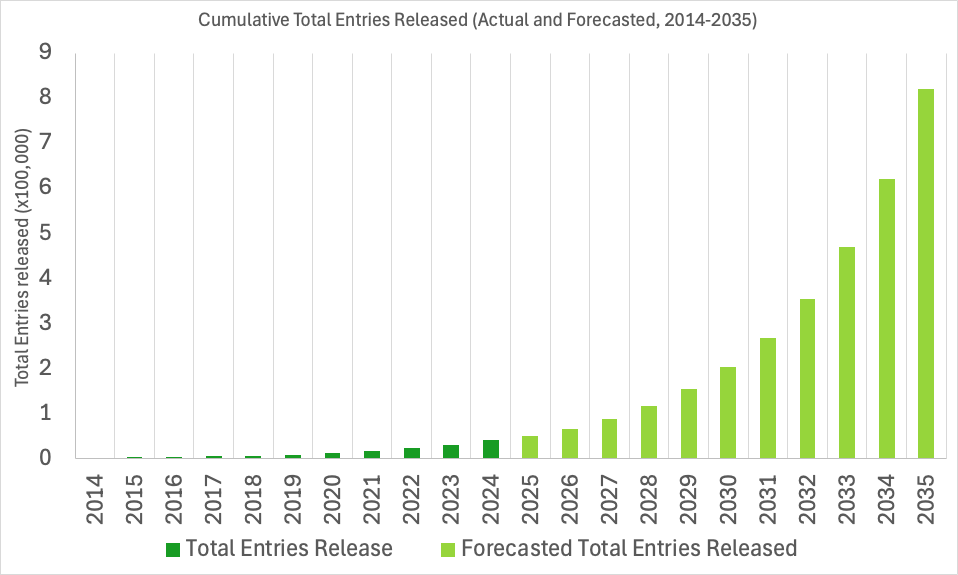 EMDB Growth
EMDB Growth
05/06/2025
Poster Prize Awarded at ASBMB
The wwPDB Foundation made an award for outstanding student presentation at the 2025 meeting of The American Society for Biochemistry and Molecular Biology (April 12–15; Chicago, IL).
 Maria Ahmed (University of Rochester) and Christine Zardecki (wwPDB Foundation)
Maria Ahmed (University of Rochester) and Christine Zardecki (wwPDB Foundation)Incorporation of methionine sulfoxides into nascent proteins during translation
Maria Ahmed, Philip Bellomio, Michael Meadow, Kevin Welle, Kyle Swovik, Jenny Hryhorenko, Sina Ghaemmaghami, University of Rochester
Many thanks to The Biophysical Society organizers and poster prize judges for making this award possible.
The wwPDB Foundation was established in 2010 to raise funds in support of the outreach activities of the wwPDB. The Foundation raised funds to help support PDB50 events, workshops, and educational publications. The Foundation is chartered as a 501(c)(3) entity exclusively for scientific, literary, charitable, and educational purposes.
The wwPDB Foundation is grateful for our industrial sponsors: Discngine and MiTeGen. Individual sponsorships are also available.
Consider supporting the next 50 years of PDB's spirit of openness, cooperation, and education with a donation to the wwPDB Foundation.
04/16/2025
Download Snapshots of the PDB Core Archive via HTTPS, SYNC, RSYNC, or FTP
Snapshots of the PDB Core archive (https://files.rcsb.org) can be downloaded via HTTPS, SYNC, RSYNC, or FTP. At RCSB PDB, AWS SYNC is supported instead of RSYNC.
Snapshots have been archived annually since 2005 to provide readily identifiable data sets for research on the PDB archive.
HTTPS Protocol
AWS SYNC
RSYNC Protocol
- PDBj (Japan): rsync -avy snapshots.pdbj.org:: .
FTP Protocol
- PDBj (Japan): ftp://snapshots.pdbj.org
03/11/2025
Poster Prize Awarded at The Biophysical Society Meeting
The wwPDB Foundation made an award for an outstanding student presentation at the 2025 Biophysical Society Meeting (February 15-19, Los Angeles, CA).
 Hsiang-Ling Huang
Hsiang-Ling HuangMechanisms of dysferlin-mediated membrane repair in health and disease
Hsiang-Ling Huang (1), Giovanna Grandinetti (1,2), Sarah M. Heissler (1), Krishna Chinthalapudi (1)
(1) Department of Physiology and Cell Biology, Dorothy M. Davis Heart and Lung Research Institute, The Ohio State University College of Medicine, United States
(2) Center for Electron Microscopy and Analysis, The Ohio State University, United States
Many thanks to The Biophysical Society organizers and poster prize judges for making this award possible.
The wwPDB Foundation was established in 2010 to raise funds in support of the outreach activities of the wwPDB. The Foundation raised funds to help support PDB50 events, workshops, and educational publications. The Foundation is chartered as a 501(c)(3) entity exclusively for scientific, literary, charitable, and educational purposes.
The wwPDB Foundation is grateful for our industrial sponsors: Discngine. Individual sponsorships are also available.
Consider supporting the next 50 years of PDB's spirit of openness, cooperation, and education with a donation to the wwPDB Foundation.
03/10/2025
New version of PDB-IHM validation reports now include assessments of crosslinking mass spectrometry-based structure models
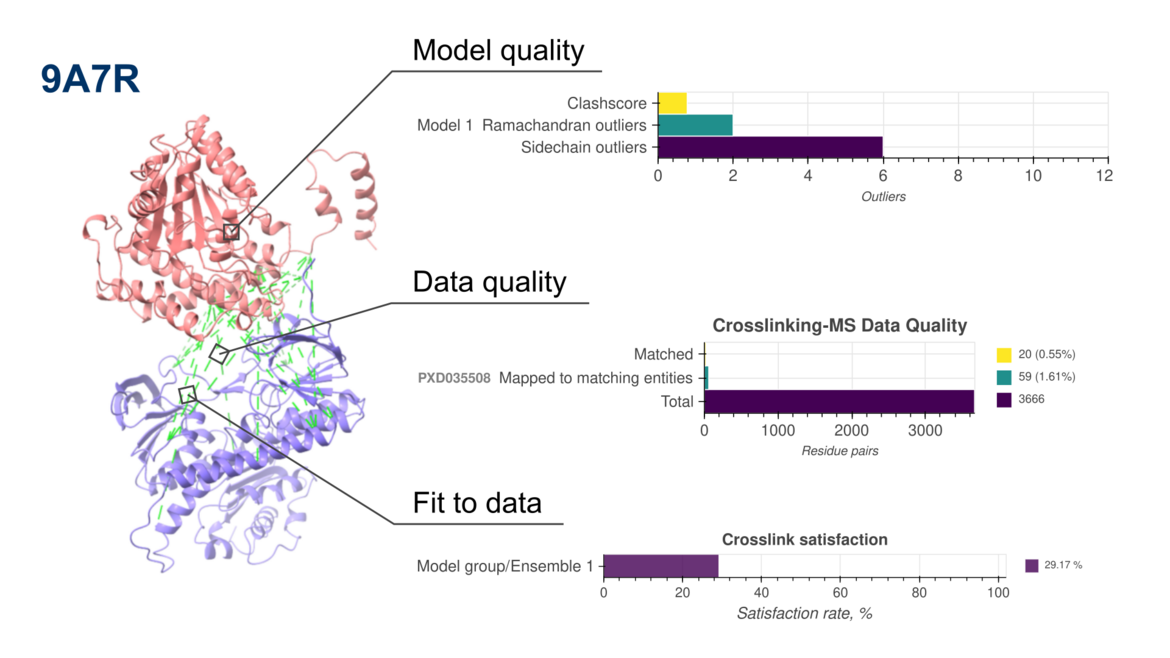 New version of PDB-IHM validation reports now include assessments of crosslinking mass spectrometry-based structure models
New version of PDB-IHM validation reports now include assessments of crosslinking mass spectrometry-based structure modelsWe are pleased to announce the release of updated validation reports (version 2) for structures determined using integrative and hybrid methods (IHM) archived in the PDB-IHM branch of the PDB archive. These reports are available in the archive and on the PDB-IHM website. IHM Validation software v2 is now integrated into the PDB-IHM deposition and curation system to support validation of new depositions. For detailed information on IHM Validation and v2 updates, please refer to the help page and user guide. The software is also available for standalone use via GitHub.
We thank the wwPDB IHM Task Force members and the broader structural biology community for supporting the development of validation methods for integrative structures.
What’s New in Version 2?
The latest update extends IHM validation to structures derived from Chemical Crosslinking Mass Spectrometry (Crosslinking-MS) data, in addition to the previously supported Small Angle Scattering (SAS) data. This milestone is the result of close collaboration among the Crosslinking-MS community, PRIDE repository, and the PDB-IHM team.
To support this advancement, we have developed interoperability mechanisms between PDB-IHM and PRIDE, enabling validation of integrative structures based on Crosslinking-MS data. Additionally, PRIDE Crosslinking now supports submission of complete Crosslinking-MS datasets following the mzIdentML 1.2.0 community data standard.
Furthermore, IHM Validation software v2 uses MolProbity version 4.5.2 and the PrISM software for precision analysis.
Next Steps for Depositors
We encourage depositors working with Crosslinking-MS-based integrative structures to:
- Submit complete datasets to PRIDE Crosslinking following the provided guidelines.
- Cross-reference PRIDE Crosslinking datasets in PDB-IHM depositions to enable robust validation.
01/29/2025
Take the PDBx/mmCIF User Guide Survey and Win!
Please take this brief survey about the PDBx/mmCIF User Guide to help us gain insights into the Guide’s usage and identify areas for future improvement.
 Benefits of the PDBx/mmCIF ecosystem
Benefits of the PDBx/mmCIF ecosystemYour feedback is highly valued.
Respondents can enter a drawing for a chance to win a prize from the wwPDB.
The survey will remain open until February 28, 2025.
01/16/2025
PDB-Dev now PDB-IHM
 Integrative structures are available at wwPDB.org and the PDB archive
Integrative structures are available at wwPDB.org and the PDB archiveStructures of many large macromolecular assemblies are now being determined using integrative approaches, wherein information derived from multiple experimental and computational methods is combined to compute their three-dimensional structures. PDB-IHM (formerly PDB-Dev) is a system for archiving and disseminating structures determined using integrative or hybrid methods (IHM), and making them Findable, Accessible, Interoperable, and Reusable (FAIR).
In August 2024, PDB-Dev was unified with the PDB to deliver integrative structures alongside experimental structures in the PDB archive. With unification, integrative structures are assigned PDB accession codes and Digital Object Identifiers (DOIs), annotated as IHM structures, and can be accessed from the PDB archive, PDB DOI links (e.g., DOI: 10.2210/pdb8zzc/pdb), and the PDB-IHM website. Now part of the PDB infrastructure, PDB-Dev has been rebranded as PDB-IHM, denoting IHM structures archived in the PDB.
Integrative structures can be deposited through the PDB-IHM deposition portal and accessible from the wwPDB OneDep home page. They are processed in parallel to the wwPDB OneDep system. Structures processed by PDB-IHM are released synchronously with PDB structures weekly on Wednesdays at 00:00 UTC.
In the future, the wwPDB partners, including Research Collaboratory for Structural Bioinformatics Protein Data Bank (RCSB PDB) in the United States, Protein Data Bank in Europe (PDBe), and Protein Data Bank Japan (PDBj), will disseminate integrative structures on their respective websites.
We look forward to supporting the structural biology community with depositing integrative structures to PDB-IHM.
Questions or feedback? Contact deposit-help@mail.wwpdb.org or heldesk@pdb-ihm.org.
01/05/2025
Time-stamped Copies of PDB and EMDB Archives
 New archive snapshots are available.
New archive snapshots are available. A snapshot of the PDB Core archive (https://files.wwpdb.org/, https://s3.rcsb.org) as of January 1, 2025 has been added to https://s3snapshots.rcsb.org/, snapshots.rcsb.org (rsync -rlpy -a -v --delete snapshots.rcsb.org:: .), and ftp://snapshots.pdbj.org. Snapshots have been archived annually since 2005 to provide readily identifiable data sets for research on the PDB archive.
The directory 20250101 includes the 229,564 experimentally-determined structure and experimental data available at that time. Atomic coordinate and related metadata are available in PDBx/mmCIF, PDB, and XML file formats. The date and time stamp of each file indicates the last time the file was modified. The snapshot of PDB Core Archive is 1,437 GB.
A snapshot of the EMDB Core archive (ftp://ftp.ebi.ac.uk/pub/databases/emdb/) as of January 01, 2025 can be found in ftp://ftp.ebi.ac.uk/pub/databases/emdb_vault/20250101/ and ftp://snapshots.pdbj.org/20250101/. The snapshot of EMDB Core Archive contains map files and their metadata within XML files for both released and obsoleted entries (41,367 and 301, respectively) and is 21 TB in size.