wwPDB Deposition Policies and wwPDB Biocuration Procedures
Section B: wwPDB Biocuration Procedures
Authored by the wwPDB annotation staff
Mar 2025 Version 4.5
Table of Contents
Preface
1. Entry title, authors, and citation information
2. Macromolecule information
3. Polymer sequences and sequence database reference assignment
4. Ligands
5. Coordinate section
6. Chain ID assignment
7. HEADER assignment
8. Assembly
9. Miscellaneous records
10. Structural Genomics Entries
11. Information specific to X-ray structures
12. Information specific to NMR structures
13. Information specific to Electron Microscopy structures
14. Viral capsids and other complex assemblies
15. Re-refinement of another author's data
Appendices:
A. HEADER list
B. Format for Structure Factors
Preface
Since 1999, the wwPDB has been responsible for processing PDB data with deposition centers at RCSB PDB, PDBe, and PDBj. The processed entries follow the mmCIF format that complies with the PDB Exchange Dictionary (PDBx)
https://mmcif.wwpdb.org/dictionaries/mmcif_pdbx_v50.dic/Index/.
The version of the PDB file and its correspondence to the file format guide will be included in all files processed and released by the wwPDB.
This document presents the current processing procedures. The wwPDB staff will continue to update annotation practices in line with evolving structure determination and annotation methods.
December 2008: Initial release as version 2.2
March 2009: minor revision 2.3, updates on citation, header list, SITE records and added cif templates for structure factors.
April 2009: minor revision 2.4, update header list.
November 2011: major revision 2.5, clarify content, update header list and UniProt sequence submission.
December 2012: Clarify Entry title.
January 2014: Clarify Re-refinement.
August 2017: Clarify Best representative conformer of NMR entries.
May 2018: Update biological assembly annotation procedure.
July 2021: major revision 4.0 to clarify content and provide examples in mmCIF format.
November 2021: IDs reserved for new ligand used in structure determination process.
November 2022: Peptide bonds in polymer chain need to be in same direction.
November 2023: Distinct chain IDs for cleaved polymer chains.
January 2024: Restriction of chain ID length
1. Entry title, authors, and citation information
Entry Title (_struct.title)
Entry titles should be descriptive and match the content of the entry. PDB staff may modify the entry title during data processing or before entry release if the title is ambiguous or does not match the content of the structure.
Authors may choose to suppress the information about the entry authors and title so that it does not appear on the PDB website until the entry is released to the public.
Author information ( _audit_author.name)
The authors for a PDB entry can be the same as the authors for the primary citation, or a subset of citation authors. Alternatively, there may be more authors listed for the entry compared to the citation author. Generally, at least one of the authors for the entry should be included in the author list for the primary citation.
The authorship of the entry is at the discretion of the principal investigator (PI). If more than one PI is responsible for the entry, they will need to come to a mutual decision on the authorship.
Citation information
Authors are encouraged to deposit their structures in advance of publication. The primary citation is the paper that describes the structure in the PDB entry.
Thesis
Conference Proceedings and Thesis can only be included as primary citation, but not in secondary citations.
PubMed Ids
PubMed IDs are available for the primary citations of entries in the PDB, mmCIF and XML files. When available, DOI numbers are also included.
Patents
Patent IDs can be recorded (_citation.pdbx_database_id_patent), and the journal name will be ‘Patent’.
Unpublished structures
If the author indicates that the entry will never be published, a journal name of 'not published' will be added to citation.journal_abbrev records.
Jr.
Journal and entry author names that have the suffix "junior" will be represented as "Jr." and not as "junior", for example, ‘Smith Jr., J.’.
Title case
All titles, names etc. are included in mixed case in the mmCIF format file to match the literature.
2. Macromolecule information
How are the macromolecule names and synonyms assigned?
Protein molecule names
Protein molecule names and synonyms are standardized to be consistent with the corresponding UniProt entry and are listed in the _entity.pdbx_description and _entity_name_com.name records. The exceptions are as follows:
- If the UniProt name refers to a precursor and the entry contains mature protein, the word precursor will not be included in the protein name. If the author did indicate putative, then the wording will be retained.
- In the case of zymogens, if the UniProt name is "trypsinogen" but the activated protein is present in the structure, then "trypsin" is used as the name.
- If the UniProt entry contains the complete gene sequence of a protein that is processed into more than one chain, the corresponding polypeptide name will be given as the name in the PDB entry. For example, if the UniProt name is insulin, the protein names correspond to the chain names: such as Insulin A chain and Insulin B chain.
- For viral proteins, where a polyprotein is synthesized, and where the PDB entry contains all the components, the name polyprotein can be used. Otherwise, the name of the fragment from UniProt will be used. For example, if a "genome polyprotein" is composed of capsid, envelope protein, and major envelope proteins but the deposited structure is only of the envelope protein, the name used will be "envelope protein".
- If the full UniProt sequence for the protein is not present in the sample, the fragment section will be filled in. However, the fragment section will not be filled in where complete mature protein is represented, for example for a complete trypsin molecule or envelope protein etc.
- If the UniProt name is not assigned (i.e., it is "hypothetical") and the author has a name for the protein, the author's protein name will be used.
- If the author's name for the molecule differs from the UniProt molecule name, the UniProt primary name will be used for the molecule name. The author's molecule name will be listed first in the synonyms list, followed by the synonyms provided in UniProt.
- If there is no corresponding UniProt entry, the author provided protein name will be used.
- Antibodies will be named using as much information as the author provides. Examples:
- Fab fragments will be named as Fab heavy chain and Fab light chain.
- For immunoglobulin G chains (which include fab and fc regions), the protein names will be "antibody heavy chain" and "antibody light chain."
- If the author provided specific names for the antibody, then those names will be used e.g., Fr62 monoclonal antibody light chain and Fr62 monoclonal antibody heavy chain.
- For chimeric proteins, the protein name is comma separated and may refer to the presence of a linker (protein_1, linker, protein_2). Other details about the chimera can be provided in entity.details and pdbx_entry_details.sequence_details.
Nucleic acid molecule names
For all nucleic acid sequences, biological names should be used when available. The biological name is either provided by the author or it is obtained from a sequence database, such as "16S RIBOSOMAL RNA".
If the sequence is shorter than 24 nucleotides and no biological name is available, the molecule name is given as the short sequence:
5'-D(*CP*GP*CP*GP*(8OG)P*AP*TP*TP*CP*GP*CP*G)-3'
For sequences equal to or greater than 24 nucleotides, when no biological name is available, the name may be listed as “50-mer RNA”, for example.
Carbohydrate polymers
As of July 2020, the rules we apply for entries containing carbohydrate polymers are the following:
(1) Residues are ordered from the reducing to non-reducing end. For common glycosylation, there is only one reducing end, with branches at non-reducing end. This is like a tree, where ordering always starts from the root (reducing end), because it's inconsistent to count from the ends of branches (non-reducing end).
(2) The longest branch is taken as main branch. If two branches have the same length, then the lower locant goes first. This is to follow 1996 IUPAC/IUBMB recommendation.
For more information, please consult https://www.wwpdb.org/documentation/carbohydrate-remediation
How is fragment information indicated? (entity.pdbx_fragment)
If the PDB entry contains a fragment of the protein in the sequence records when compared to the UniProt entry, the fragment name can be included, for example: N-terminal domain, C-terminal domain, catalytic domain, ligand binding domain, etc.
How is the E.C. number assigned (entity.pdbx_ec)?
The E.C. number is automatically extracted from the UniProt entry. If the UniProt entry does not have an E.C. number information, the author’s provided information will be used. If the author disagrees with the UniProt assignment of E.C. number, the wwPDB staff will contact the UniProt staff and try to clarify the discrepancy. In the case where the matter cannot be resolved, the author provided E.C. information can be added to the entity.details and indicated as "Author provided E.C. number is xxxx".
How are mutations indicated (_entity.pdbx_mutation)?
The exact mutation will be described in the mmCIF file in the following manner: Y20K. The sequence difference records (struct_ref_seq_dif) will contain a corresponding record with the annotation "ENGINEERED MUTATION".
How is the macromolecule source organism indicated?
The source organism is identified by its scientific name and taxonomy id as listed in the NCBI Taxonomy database. During entry annotation the official scientific name is usually obtained from UniProt database which is based on the NCBI Taxonomy database. The source organism name and taxonomy id will be listed under _entity_src_nat mmCIF category if the polymer molecule was naturally obtained, under _entity_src_man if it was genetically manipulated (expressed) and under _pdbx_entity_src_syn if it was chemically synthesized. If a common name is listed in the NCBI taxonomy database, then the common name is mapped to the _entity_src_gen.gene_src_common_name field, otherwise this will be left blank. The scientific name of the source and host organisms, plasmid and gene names will be included in mixed case to match the standard scientific literature.
The source organism names of each part of a chimeric protein are listed in the mmCIF files with corresponding residue ranges.
For example:
loop_
_entity_src_gen.entity_id
_entity_src_gen.pdbx_src_id
_entity_src_gen.pdbx_alt_source_flag
_entity_src_gen.pdbx_seq_type
_entity_src_gen.pdbx_beg_seq_num
_entity_src_gen.pdbx_end_seq_num
_entity_src_gen.gene_src_common_name
_entity_src_gen.gene_src_genus
_entity_src_gen.pdbx_gene_src_gene
_entity_src_gen.gene_src_species
_entity_src_gen.gene_src_strain
_entity_src_gen.gene_src_tissue
_entity_src_gen.gene_src_tissue_fraction
_entity_src_gen.gene_src_details
_entity_src_gen.pdbx_gene_src_fragment
_entity_src_gen.pdbx_gene_src_scientific_name
_entity_src_gen.pdbx_gene_src_ncbi_taxonomy_id
_entity_src_gen.pdbx_gene_src_variant
_entity_src_gen.pdbx_gene_src_cell_line
_entity_src_gen.pdbx_gene_src_atcc
_entity_src_gen.pdbx_gene_src_organ
_entity_src_gen.pdbx_gene_src_organelle
_entity_src_gen.pdbx_gene_src_cell
_entity_src_gen.pdbx_gene_src_cellular_location
_entity_src_gen.host_org_common_name
_entity_src_gen.pdbx_host_org_scientific_name
_entity_src_gen.pdbx_host_org_ncbi_taxonomy_id
_entity_src_gen.host_org_genus
_entity_src_gen.pdbx_host_org_gene
_entity_src_gen.pdbx_host_org_organ
_entity_src_gen.host_org_species
_entity_src_gen.pdbx_host_org_tissue
_entity_src_gen.pdbx_host_org_tissue_fraction
_entity_src_gen.pdbx_host_org_strain
_entity_src_gen.pdbx_host_org_variant
_entity_src_gen.pdbx_host_org_cell_line
_entity_src_gen.pdbx_host_org_atcc
_entity_src_gen.pdbx_host_org_culture_collection
_entity_src_gen.pdbx_host_org_cell
_entity_src_gen.pdbx_host_org_organelle
_entity_src_gen.pdbx_host_org_cellular_location
_entity_src_gen.pdbx_host_org_vector_type
_entity_src_gen.pdbx_host_org_vector
_entity_src_gen.host_org_details
_entity_src_gen.expression_system_id
_entity_src_gen.plasmid_name
_entity_src_gen.plasmid_details
_entity_src_gen.pdbx_description
1 1 sample 'Biological sequence' 1 121 ? ? ? ? ? ? ? ? ? 'Mus musculus' 10090 ? ? ? ? ? ? ? ? 'Escherichia coli' 562 ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ?
1 2 sample 'Biological sequence' 122 172 ? ? ? ? ? ? ? ? ? 'Homo sapiens' 9606 ? ? ? ? ? ? ? ? 'Escherichia coli' 562 ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ?
If an expression system was used, the scientific organism name of the expression system will also be taken from the NCBI Taxonomy database and listed under _entity_src_gen.pdbx_host_org_ncbi_taxonomy_id records.
Other information about the source and expression system is not mandatory and will be included in the file only if the author has provided it.
Phage display
The _entity_src_gen category will be populated with the organism where the gene sequence was first isolated and the host organism of the overexpression system used to generate the final protein. Additional information about phage display can be mapped to _entity_src_gen.pdbx_description for example: ’Protein selected by phage display’.
Cell-free synthesis, in vitro transcription and in vitro translation
Cell-free synthesis, in vitro transcription and in vitro translation will all be described as "cell-free synthesis". Common cell extracts in use today for cell-free protein production are made from E. coli (ECE), rabbit reticulocytes (RRL), wheat germ (WGE), insect cells (ICE) and Yeast Kluyveromyces (the D2P system). All will have genetically manipulated sources (_entity_src_gen category) populated with the above-mentioned source organisms and the relevant taxonomy ids. Other information about the synthesis, such as "cell-free synthesis" can be added to the _entity_src_gen.pdbx_description.
Baculovirus
If a baculovirus was used, it would be listed under the vector type. If a cell line is provided, it will be added to the entry.
Synthetic
The term "synthetic" means "synthesized using non biological methods". Taxonomy ID for synthetic source is 32630.
Undefined
This term is used for cases when the source organism cannot be identified. Taxonomy ID for undefined source is 32644.
3. Polymer sequences and sequence database reference assignment
What is the definition of polymer sequence (entity_poly, SEQRES)?
The polymer sequence is a list of the consecutive chemical components covalently linked in a linear fashion to form a polymer. The chemical components included in this listing may be standard or modified amino acid or nucleic acid residues. It may also include other residues that are linked to the standard backbone in the polymer. Chemical components or groups covalently linked to the side chains of peptides or groups linked to sugars and/or bases in nucleic acid polymers will not be listed here.
Proteins containing three or more residues, forming two consecutive standard peptide bonds, in the same direction will be assigned polymer sequence records (molecule name, source, sequence, sequence database reference).
Nucleic acids containing two or more residues linked by standard nucleotide bonds will be assigned polymer sequence records (molecule name, source, sequence, sequence database reference).
The polymer sequence records must represent the complete sequence of each sample used in the experiment, including any expression tags, and residues not modelled in the coordinates (for example unobserved regions due to local disorder). Residues cleaved from the polymer sequence prior to or during the experiment are not considered part of the sequence. The polymer sequence can include neighboring cross-linked residues (such as chromophores) and modified amino acids.
What if the exact sequence of the sample is not known?
If the exact sequence of the sample is not known, due to, for example, proteolysis, the sequence should match the coordinates and a dedicated sequence remark (_pdbx_entry_details.sequence_details, REMARK 999) with an explanation should be added. In such cases if the entry is a crystal structure, Matthew's coefficient and solvent content will list author-provided values instead of calculated values.
What is the sequence database reference (_struct_ref, _struct_ref_seq, DBREF)?
The sequence database reference provides the mapping between polymer sequences and a valid sequence database reference.
Which polymer chains are assigned sequence database records?
Each protein or nucleic acid chain, for which there is an appropriate sequence database match will list cross references to the sequence database entry. When no sequence database reference is available the sequence will be self-referenced (i.e., the database reference will be the PDB entry itself).
Which sequence databases will be used?
- Proteins
UniProt is the current preferred protein sequence database. Where there are multiple UniProt entries for the same protein from the same organism, strain and sequence identity, the UniProt entry that has the most annotation will be used. Also, UniProt entries that contain the complete protein sequence will be preferred over those that represent protein fragments.
- Nucleic acids
Naturally obtained nucleic acid sequences can be referenced to GenBank.
- Non-ribosomal peptides
NORINE database was used for some sequence database references. The reference to NORINE is currently discontinued. UniProt reference (if available) or self-reference is used instead.
- DNA and RNA
All DNA will be self-referenced. RNA polymers more than 50 residues long, will be provided GenBank sequence reference if available.
What if a UniProt reference is not available for a protein sequence?
UniProt does not contain variable or hyper-variable regions of the immune system or unnatural sequences, so the PDB entries for such structures will be self-referenced. If the protein does not fall into these categories and does not have a UniProt reference, UniProt automatically obtains sequences from the PDB and adds them to UniProt.
What if a UniProt reference has become available or has been changed in the UniProt database for a protein sequence?
The PDB entry contains the sequence cross reference available at the time of processing of the entry. If at a point in the future the sequence does appear in UniProt or if a UniProt accession code has been changed, the updated version of the sequence database match will be available to users through the SIFTS project. The PDB entry may not be modified to add/update the sequence cross-reference. Information about SIFTS is available from https://www.ebi.ac.uk/pdbe/docs/sifts/
If related entries with the same sequence do not have UniProt references, the sequence reference will refer to each PDB entry itself and not refer to the first PDB entry which contained the particular sequence. For example, if entries 1ABC, 1DEF and 1GHI all have the same antibody sequence, these entries will refer to 1ABC, 1DEF and 1GHI, respectively.
How are chimeras handled?
Chimeras or fusion proteins should be deposited as a single chain with one chain ID because they were expressed as one chain. Chimeras that were refined with different chain IDs should be deposited with one chain ID for all parts of the chimera, including any linker regions. The sections of the chimera which match the UniProt entry or entries will each have database references. The sections of the chimera which do not match the UniProt entry or entries, will be self-referenced.
What is the sequence reference difference (_struct_ref_seq_dif, SEQADV)?
The sequence difference records describe any rational disagreement between a sequence database and the sequence in the PDB entry.
What are the various types of sequence differences and how are they annotated?
- Engineered mutation:
Difference between the PDB sequence and the UniProt entry that were engineered will be listed as “engineered mutation”.
- Cloning artifact:
The term cloning artifact is reserved for instances where a sequence difference is introduced, as in a PCR experiment during cloning or by random mutation. These instances are rare.
- Modified residue:
Only instances where the parent of a modified residue does not match the sequence database reference will be listed here. For example, if THR is listed in the UniProt entry, and the PDB residue is MSE (selenomethionine), then struct_ref_seq_dif (SEQADV) record for MSE/THR with the explanation of "engineered mutation" will be generated, all other cases of modified residues will be listed under pdbx_struct_mod_residue (MODRES) records.
- Microheterogeneity:
See section “How is microheterogeneity/polymorphism handled?” below.
- Chromophore:
See section "Neighboring intra-chain cross-linked groups" below.
- Conflict:
Sequence conflicts between a residue in the polymer sequence and that listed in the UniProt reference that cannot otherwise be explained, will be listed here with the explanation of "conflict" and additional sequence details may also be provided.
- Expression tag:
Extra N- and/or C-terminal residues including leader sequences, His-tags, other kinds of tags and/or initiating methionine(s).
- Insertion:
in the middle of the sequence that is not part of the UniProt sequence
- Deletion:
where the construct of the protein in the middle of sequence that has the UniProt reference is deleted.
- Linker:
a region in sample sequence linking partitions of a chimeric entity.
What sequence differences are not listed?
- Polymers which do not have a UniProt reference (self-referenced).
- Modified residues where the parent residue matches the database reference.
MODRES records (_pdbx_struct_mod_residue) will be created for modified residues if the residue is derived from a parent residue. For instance, MSE (selenomethionine) will only have a pdbx_struct_mod_residue (MODRES) entry when Met is listed in the UniProt entry.
- D-amino acids
How is microheterogeneity/polymorphism handled?
Polymer chains which have the same sequence must have the same chemistry (homogeneous). If one chain has microheterogeneity at one position, but not in another chain at the same position, then these two polymer chains will be treated as two different polymer entities with two sequences. If a residue has more than one identity at a particular position within a chain, this is called microheterogeneity.
For heterogeneous chains, the residue which does not match the corresponding UniProt residue will be listed in the sequence, regardless of its occupancy. The total sum of occupancies of the different identities of the residues that display microheterogeneity should be less than or equal to 1.
If none of the residue identities match the UniProt residue at the corresponding sequence location, the residue with the higher occupancy will be listed in the sequence. A sequence difference record will be generated for the difference between the UniProt sequence and the polymer sequence, with the explanation of "microheterogeneity". If there is no UniProt reference, the higher occupancy residue will be listed in the sequence and no sequence difference records will be generated.
If a residue has two identities at a particular residue number, where one identity is a modified residue and one is the unmodified form, then the modified residue will be listed in the polymer sequence, and a "microheterogeneity" record will be generated in the sequence difference category. This is the exception to the rule that sequence difference records would not be created for modified residues.
Alternate position indicators will be used in the coordinates for residues involved in microheterogeneity. The residue listed in the polymer sequence will be listed first in the coordinates and be labeled as alternate position identifier A. The other identity will be assigned alternate position identifier B. If there is a third or fourth residue identity, or if one of the identities has its own alternate conformations, these will be assigned alternate IDs in alphabetical order.
In the mmCIF file, all residue identities involved in microheterogeneity and a microheterogeneity flag will be listed in _entity_poly_seq.hetero and _poly_seq_scheme.hetero.
Example (PDB entry 5ZA2):
loop_
_entity_poly_seq.entity_id
_entity_poly_seq.num
_entity_poly_seq.mon_id
_entity_poly_seq.hetero
...
2 56 ILE n 2 57 GLY n 2 58 SEP y
2 58 SER y 2 59 VAL n 2 60 SER n
...
loop_
_pdbx_poly_seq_scheme.asym_id
_pdbx_poly_seq_scheme.entity_id
_pdbx_poly_seq_scheme.seq_id
_pdbx_poly_seq_scheme.mon_id
_pdbx_poly_seq_scheme.ndb_seq_num
_pdbx_poly_seq_scheme.pdb_seq_num
_pdbx_poly_seq_scheme.auth_seq_num
_pdbx_poly_seq_scheme.pdb_mon_id
_pdbx_poly_seq_scheme.auth_mon_id
_pdbx_poly_seq_scheme.pdb_strand_id
_pdbx_poly_seq_scheme.pdb_ins_code
_pdbx_poly_seq_scheme.hetero
...
B 2 56 ILE 56 62 62 ILE ILE B . n
B 2 57 GLY 57 63 63 GLY GLY B . n
B 2 58 SEP 58 64 64 SEP SEP B . y
B 2 58 SER 58 64 64 SER SER B . y
B 2 59 VAL 59 65 65 VAL VAL B . n
B 2 60 SER 60 66 66 SER SER B . n
...
A full explanation of the microheterogeneity for all residues at a particular residue number may be further elaborated with additional sequence details text remark.
Neighboring intra-chain cross-linked groups
This section describes cases of neighboring intra-chain cross-linked typically involving 2 or more sequential amino acids that react with each other to form one "residue". For example, residues 65, 66, and 67 in the PDB entry 1yjf underwent a reaction to form a circularized tri-peptide chromophore (Figure 1).
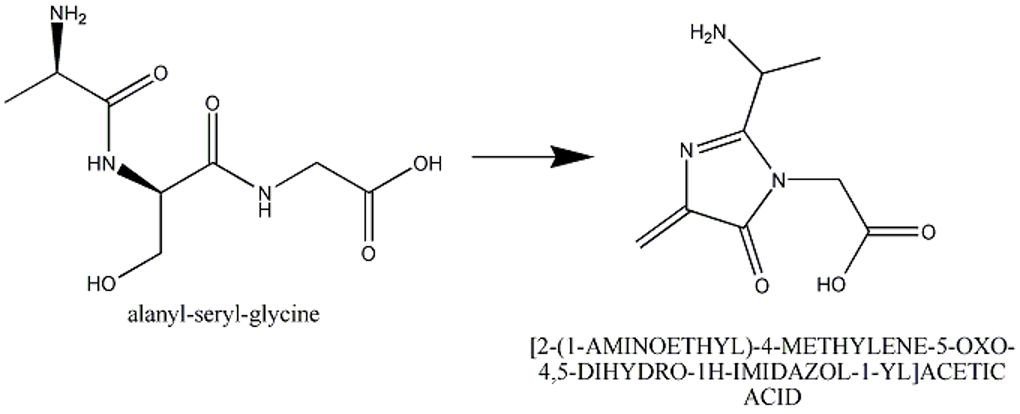
Figure 1: In entry 1yjf, residues 65, 66, and 67 (neighboring alanine, serine and glycine residues) reacted and generated a chromophore product called [2-(1-AMINOETHYL)-4-METHYLENE-5-OXO-4,5-DIHYDRO-1H-IMIDAZOL-1-YL]ACETIC ACID.
To describe this situation in a PDB entry the following annotation applies:
- The neighboring intra-chain cross-linked group will be listed as one chemical group in the coordinates and in the sequence.
- The neighboring intra-chain cross-linked group will have three parent residues, if three amino acids were involved in the reaction to make the chromophore. Thus, there will be three pdbx_struct_mod_residue (MODRES) records.
- There will be struct_ref_seq_dif (SEQADV) record(s) generated with the explanation "chromophore". Any additional information can be added to pdbx_entry_details.sequence_details (REMARK 999).
4. Ligands
The wwPDB encourages depositors to provide an InChI or SMILES string and/or chemical name and/or chemical drawing including bond type, bond order and stereochemistry of the ligand in order to facilitate the correct annotation.
How is the identity of a ligand verified?
Specialized software is used to get bond types, stereochemistry, and where possible the IUPAC-compliant name for each ligand in the structure. Verification is done by comparing to the ligand structure in the coordinates vs the one defined in the wwPDB Chemical Component Dictionary (CCD). When a ligand match is found in the CCD, the atom names in the coordinates are matched to the dictionary definition.
How is the ligand identity assigned?
Each component (ligand, amino acid, nucleic acid) is assigned a unique code in the CCD. During curation, annotators use multiple methods to compare components against the CCD. If stereochemistry, bond types and connectivity match an existing definition in the CCD then the existing definition is assigned to the ligand. If there is no match, a new CCD definition is added to the CCD with an arbitrarily assigned unique identifier.
How are new ligands added to the chemical component dictionary?
To designate the new ligands we encourage the use of the following ids in the structure determination process: DRG, LIG, INH, 00 - 99 (2 digits). These ids are excluded from the CCD, and are instead reserved for initial deposition of any ligand that is new to the CCD. Several software programs are used to generate a CCD definition based on the author's deposited coordinates. All atoms in the component CIF file (including H-atoms) will have coordinates. If there are atoms missing from the coordinates due to disorder, they will be added to the component CIF file. A set of ideal coordinates and an IUPAC-compliant chemical name will be generated for the complete molecule. Checks for chemical name, ligand code and connectivity are carried out to verify that duplicates are not added to the CCD.
What happens if a structure contains a ligand that already exists in the chemical component dictionary?
If a particular ligand in the structure is already defined in the CCD, the ligand 3-letter identifier, its chemical name and atom nomenclature will be updated according to the dictionary during entry annotation. It is noted that the atom names will always start with the element type.
Are peptide-like inhibitors treated as ligands?
If a peptide-like inhibitor is a natural product or if it has a sequence database reference, or if the majority of the residue components are standard or modified amino acids, it is treated as a polymer. Otherwise, the molecule is treated as a ligand.
Charge state
Whenever possible, the overall charge for new ligand definitions should be neutral. This provides maximum compatibility with other chemical databases such as CAS and PubChem. The overall charge of the ligand is included in the CCD. The individual atoms may have a charge in the atom records.
Exceptions:
- Tetravalent nitrogen atoms where all four bonding partners are heavy atoms must carry a positive charge to satisfy valence rules
- Nitro groups (R-NO2) are represented in a charge separated state (R-[N+](=O)[O-]), again, to better satisfy valence rules.
- Organometallic complexes
Ligands in NMR structures produce unique challenges because all hydrogen atoms used in the structure must be present in the dictionary. The accurate chemical description of molecules which may exist in multiple protonation states is difficult to achieve in single chemical component definitions. To describe this complexity for the standard amino acids and nucleotides, a special component dictionary has been created (https://www.wwpdb.org/data/ccd). Within this dictionary are complete chemical descriptions of observed protonation states which include accurate formula, formal charges and IUPAC atom nomenclature for each case.
How are ligand names and synonyms assigned?
_chem_comp.name (HETNAM) Whenever possible, the name automatically produced by specialized chemical naming software is used. Exceptions to this rule are common (as judged by annotation staff) biological names, and brand names for drugs. Should ACDlabs, Chemdraw or PubChem fail to predict a name, common names or names supplied by the depositor may be used.
_chem_comp.pdbx_synonyms, _pdbx_chem_comp_synonyms (HETSYN) Other names requested by depositors may be included as synonyms (_pdbx_chem_comp_synonyms), at the discretion of the annotator. It should be noted any synonyms provided by the depositor should be meaningful and widely used names for the ligand.
Exceptions / Issues: Established common names can be used as molecule names as long as the IUPAC-compliant name is listed as a synonym name. For example, "Fluconazole" can be used as molecule name and "2-(2,4-difluorophenyl)-1,3-bis(1,2,4-triazol-1-yl)propan-2-ol" as a synonym. If a ligand name is changed or synonyms are added, every PDB entry containing that ligand will be updated.
Metals: There are difficulties in handling dative bonds in coordination complexes and pi-bonding in organometallic complexes. These inorganic molecules need to be built on a case-by-case basis to reflect their chemical nature. An ambiguous flag (_chem_comp.pdbx_ambiguous_flag) is included in the chemical component files for such molecules where current cheminformatics software is inadequate or unable to describe the true chemical structure.
A number of ligands that were defined as "metal bound to multiple waters" have been marked as obsolete (status code of OBS) meaning they are no longer valid ligands, and should not be used in the future. While water coordinated metals can be strongly bound, these groups are not consistently used among all structures, and user analysis would benefit from standardizing the single ion representation. Entries which contained such ligands were updated to split them into metal and water molecules.
Leaving groups: In order to accurately represent the polymerization potential of entries in the chemical component dictionary, all atoms that can be lost from a ligand in the process of making a linkage will be marked with a special flag indicating these are 'leaving atoms'. For example, standard amino acids have the carbonyl oxygen atom OXT, and the N-terminal hydrogen HN2 are always marked as leaving atoms as these are lost in the process of creating a polypeptide chain. This, however, does not suggest that these atoms are always missing from the coordinates, but rather these atoms are invariably missing in a polymeric state depending on the position and bonding state of the chemical entity.
Portions of ligands that are missing: Ligands are defined as if all atoms were present in the experiment. If the ligand is only partially seen in the experiment (for example, a ring is missing in the density for a crystal structure), the ligand code that is used is for the fully defined ligand. If the ligand is new, the missing atoms are added to the definition of the ligand.
Modified amino acids and nucleotides: If an amino acid or nucleotide is modified by a chemical group greater than 10 atoms, the residue will be split into two groups: the amino acid/nucleotide group and the modification. A link record will be generated between the amino acid/nucleotide group and the modification. Modified amino acids and nucleotides will follow standard atom nomenclature.
Nucleotide residue identifiers: The standard nucleotides are represented using the following residue identifiers for DNA: DA, DG, DC, DT and the following residue identifiers for RNA: A, G, C, U.
Use of UNX/UNL/UNK There are times when an amino acid residue, nucleotide, atom, or ligand is unidentified. These ligand codes should be used in the following cases:
- UNX: unknown atom or ion
- UNL: unknown ligand
- UNK: unknown amino acid
- N/DN: unknown nucleotide
UNX UNX is the code for one atom or ion, by itself, when author does not know the identity of that atom or ion. NOTE: The ligand name is UNX, but the atom name is UNK. The atom type is "X".
UNL UNL is the code for unknown ligands. This is for where author has added atoms to the coordinates to satisfy the electron density, but true ligand identity is not known. For example, see PDB entry 3MHO.
UNK UNK is the code for unknown amino acids only. For example, a poly-ALA or poly-GLY chain would be processed as poly-UNK, if the author does not know how the coordinates align with the sequence and the residue numbering is arbitrary. The sequence would be poly UNK and the residues in coordinates would be listed as UNK. The sequence, if known, may be listed in the sequence details section. If the authors do know the alignment of sequence and coordinates, the poly-ALA or poly-GLY residues should be changed to match the sequence. The atom names of UNK are N,CA,CB,CG,O,C, and the atom types are N,C,C,C,O,C.
N/DN is the code for unknown nucleotide where the base is not observed.
What if the ligand identity provided by the depositor conflicts with the software and annotator identification of a ligand?
Conflicts between the author's identification of a ligand and the software and/or annotator identification of a ligand are brought to the attention of the author. In case of disagreement between the depositor and the wwPDB staff, the ligand name will be based on what is derived from the coordinates by specialized chemical naming software. If stereochemistry cannot be determined by a program, the author's description of stereochemistry will be used. For ambiguous stereochemistry or bond length, _pdbx_entry_details.nonpolymer_details (REMARK 600) and a flag might be added to the mmCIF file.
The Biologically Interesting Molecule Reference Dictionary (BIRD)
The Biologically Interesting molecule Reference Dictionary (BIRD) contains information about biologically interesting peptide-like antibiotic and inhibitor molecules in the PDB archive.
BIRD is an external reference file (similar to the CCD) that provides information about the chemistry, biology, and structure of these molecules.
BIRD entries include molecular weight and formula, polymer sequence and connectivity, descriptions of structural features and functional classification, natural source (if any), and external references to corresponding UniProt or Norine entries.
The entire BIRD resource can be downloaded from the wwPDB FTP area:
BIRD is regularly reviewed for consistency and accuracy, and is used to uniformly annotate PDB entries containing these molecules. The dictionary is updated each week with new definitions as the corresponding PDB entries are released in the PDB archive.
The corresponding BIRD ID code only appears in the PDBx-formatted file of the entry. Additional details about BIRD annotation are available at: https://www.wwpdb.org/data/bird
5. Coordinate section
Alternate conformations
How are alternate conformations of individual atoms, side chains, or entire residues handled?
Sometimes an atom, several atoms, or an entire side chain has more than one conformation. Each set of coordinates for the atom is assigned alternate position A and B, or if there are three alternate positions, A, B, and C, etc. The combined occupancies of the alternate positions should not exceed 1.00. Generally alternate conformation A should have the higher occupancy.
Example
loop_
_atom_site.group_PDB
_atom_site.id
_atom_site.type_symbol
_atom_site.label_atom_id
_atom_site.label_alt_id
_atom_site.label_comp_id
_atom_site.label_asym_id
_atom_site.label_entity_id
_atom_site.label_seq_id
_atom_site.pdbx_PDB_ins_code
_atom_site.Cartn_x
_atom_site.Cartn_y
_atom_site.Cartn_z
_atom_site.occupancy
_atom_site.B_iso_or_equiv
_atom_site.pdbx_formal_charge
_atom_site.auth_seq_id
_atom_site.auth_comp_id
_atom_site.auth_asym_id
_atom_site.auth_atom_id
_atom_site.pdbx_PDB_model_num
...
ATOM 1179 C CA A GLU A 1 75 ? 3.108 5.119 18.896 0.58 12.70 ? 167 GLU A CA 1
ATOM 1180 C CA B GLU A 1 75 ? 3.163 5.208 18.892 0.42 12.51 ? 167 GLU A CA 1
…
How are alternate conformations of chemical groups handled?
Chemical group alternate conformations are handled in the same way as amino acids, unless the alternate conformations involve two different chemical groups.
What if two different chemical groups are in alternate conformations?
The author may state that two chemical groups are alternate conformations of each other but have different identities. For example, the author may state that the chemical group at a particular location is both zinc and copper. Unlike polymorphic residues, ligands cannot be assigned the same residue number; different residue numbers must be assigned. In this example, zinc would be residue 100, and copper residue 101. Zinc would be assigned
alternate conformation A, and copper would be assigned alternate conformation B. The ligand with the higher occupancy is generally, but not always, assigned alternate conformation A. The occupancies of each ligand should be less than 1.00 and combined should be less than or equal to 1.00.
Example
loop_
_atom_site.group_PDB
_atom_site.id
_atom_site.type_symbol
_atom_site.label_atom_id
_atom_site.label_alt_id
_atom_site.label_comp_id
_atom_site.label_asym_id
_atom_site.label_entity_id
_atom_site.label_seq_id
_atom_site.pdbx_PDB_ins_code
_atom_site.Cartn_x
_atom_site.Cartn_y
_atom_site.Cartn_z
_atom_site.occupancy
_atom_site.B_iso_or_equiv
_atom_site.pdbx_formal_charge
_atom_site.auth_seq_id
_atom_site.auth_comp_id
_atom_site.auth_asym_id
_atom_site.auth_atom_id
_atom_site.pdbx_PDB_model_num
...
HETATM 2239 C C A CO2 C 3 . ? -6.625 1.156 17.412 0.33 6.76 ? 301 CO2 A C 1
HETATM 2240 O O1 A CO2 C 3 . ? -7.570 1.048 16.676 0.33 7.80 ? 301 CO2 A O1 1
HETATM 2241 O O2 A CO2 C 3 . ? -5.717 1.284 18.182 0.33 7.89 ? 301 CO2 A O2 1
HETATM 2242 C C B BCT D 4 . ? -5.601 0.708 16.643 0.67 9.63 ? 303 BCT A C 1
HETATM 2243 O O1 B BCT D 4 . ? -6.583 1.348 16.099 0.67 12.22 ? 303 BCT A O1 1
HETATM 2244 O O2 B BCT D 4 . ? -5.469 -0.535 16.378 0.67 9.77 ? 303 BCT A O2 1
HETATM 2245 O O3 B BCT D 4 . ? -4.819 1.444 17.442 0.67 12.28 ? 303 BCT A O3 1
…
Handling of terminal atoms that should not be included
Terminal atoms (OXT, HXT, H1, H2 and H3 atoms for proteins, OP3, HOP3, HO3 and HO5 atoms for nucleic acids) should not be included for the residues which are not terminal residues of the sequence, as this does not describe the true content of the crystal and is chemically incorrect. If such atoms are present in the chain, the wwPDB will remove them.
Missing residues (_pdbx_unobs_or_zero_occ_residues, REMARK 465) and atoms (_pdbx_unobs_or_zero_occ_atoms, REMARK 470)
Missing residues (for example: at the N- or C-termini or in flexible loops) and missing side chain atoms of the polymer component are listed in REMARK 465 if listed in _pdbx_unobs_or_zero_occ_residues with polymer_flag (Y) and occupancy_flag (1).
Similarly, missing atoms are listed in REMARK 470 if listed in _pdbx_unobs_or_zero_occ_atoms with polymer_flag (Y) and occupancy_flag (1). Atoms which are leaving atoms such as polymer linkage (OXT in amino acids, OP3 in nucleic acids, O1 in saccharides) and hydrogens will not be listed as missing.
Example
For missing residues
loop_
_pdbx_unobs_or_zero_occ_residues.id
_pdbx_unobs_or_zero_occ_residues.PDB_model_num
_pdbx_unobs_or_zero_occ_residues.polymer_flag
_pdbx_unobs_or_zero_occ_residues.occupancy_flag
_pdbx_unobs_or_zero_occ_residues.auth_asym_id
_pdbx_unobs_or_zero_occ_residues.auth_comp_id
_pdbx_unobs_or_zero_occ_residues.auth_seq_id
_pdbx_unobs_or_zero_occ_residues.PDB_ins_code
_pdbx_unobs_or_zero_occ_residues.label_asym_id
_pdbx_unobs_or_zero_occ_residues.label_comp_id
_pdbx_unobs_or_zero_occ_residues.label_seq_id
1 1 Y 1 A MET 1 ? A MET 1
2 1 Y 1 A ARG 2 ? A ARG 2
For missing atoms
loop_
_pdbx_unobs_or_zero_occ_atoms.id
_pdbx_unobs_or_zero_occ_atoms.PDB_model_num
_pdbx_unobs_or_zero_occ_atoms.polymer_flag
_pdbx_unobs_or_zero_occ_atoms.occupancy_flag
_pdbx_unobs_or_zero_occ_atoms.auth_asym_id
_pdbx_unobs_or_zero_occ_atoms.auth_comp_id
_pdbx_unobs_or_zero_occ_atoms.auth_seq_id
_pdbx_unobs_or_zero_occ_atoms.PDB_ins_code
_pdbx_unobs_or_zero_occ_atoms.auth_atom_id
_pdbx_unobs_or_zero_occ_atoms.label_alt_id
_pdbx_unobs_or_zero_occ_atoms.label_asym_id
_pdbx_unobs_or_zero_occ_atoms.label_comp_id
_pdbx_unobs_or_zero_occ_atoms.label_seq_id
_pdbx_unobs_or_zero_occ_atoms.label_atom_id
1 1 Y 1 B ILE 137 ? CG1 ? B ILE 137 CG1
2 1 Y 1 B ILE 137 ? CG2 ? B ILE 137 CG2
3 1 Y 1 B ILE 137 ? CD1 ? B ILE 137 CD1
Zero occupancy residues
Some refinement programs allow inclusion of missing residues and side chain atoms in the coordinate files as atoms with occupancy 0.00. Since these atoms are usually ignored during refinement, their location and properties may not be reliable. Therefore, if such atoms are included in the deposited coordinate file, these atoms will be retained in the file but also listed in separate remarks: REMARK 475 if listed in _pdbx_unobs_or_zero_occ_residues with polymer_flag (Y) and occupancy_flag (0) (for zero occupancy residues) and REMARK 480 if listed in pdbx_unobs_or_zero_occ_atoms with polymer_flag (Y) and occupancy_flag (0). Atoms which are leaving atoms such as polymer linkage (OXT in amino acids, OP3 in nucleic acids, O1 in saccharides) and hydrogens will not be listed as missing.
For zero occupancy residues:
loop_
_pdbx_unobs_or_zero_occ_residues.id
_pdbx_unobs_or_zero_occ_residues.PDB_model_num
_pdbx_unobs_or_zero_occ_residues.polymer_flag
_pdbx_unobs_or_zero_occ_residues.occupancy_flag
_pdbx_unobs_or_zero_occ_residues.auth_asym_id
_pdbx_unobs_or_zero_occ_residues.auth_comp_id
_pdbx_unobs_or_zero_occ_residues.auth_seq_id
_pdbx_unobs_or_zero_occ_residues.PDB_ins_code
_pdbx_unobs_or_zero_occ_residues.label_asym_id
_pdbx_unobs_or_zero_occ_residues.label_comp_id
_pdbx_unobs_or_zero_occ_residues.label_seq_id
1 1 Y 0 A PRO 38 ? A PRO 1
2 1 Y 0 A HIS 39 ? A HIS 2
For zero occupancy atoms:
loop_
_pdbx_unobs_or_zero_occ_atoms.id
_pdbx_unobs_or_zero_occ_atoms.PDB_model_num
_pdbx_unobs_or_zero_occ_atoms.polymer_flag
_pdbx_unobs_or_zero_occ_atoms.occupancy_flag
_pdbx_unobs_or_zero_occ_atoms.auth_asym_id
_pdbx_unobs_or_zero_occ_atoms.auth_comp_id
_pdbx_unobs_or_zero_occ_atoms.auth_seq_id
_pdbx_unobs_or_zero_occ_atoms.PDB_ins_code
_pdbx_unobs_or_zero_occ_atoms.auth_atom_id
_pdbx_unobs_or_zero_occ_atoms.label_alt_id
_pdbx_unobs_or_zero_occ_atoms.label_asym_id
_pdbx_unobs_or_zero_occ_atoms.label_comp_id
_pdbx_unobs_or_zero_occ_atoms.label_seq_id
_pdbx_unobs_or_zero_occ_atoms.label_atom_id
1 1 Y 1 A GLN 14 ? CG ? A GLN 16 CG
2 1 Y 1 A GLN 14 ? CD ? A GLN 16 CD
3 1 Y 1 A GLN 14 ? OE1 ? A GLN 16 OE1
4 1 Y 1 A GLN 14 ? NE2 ? A GLN 16 NE2
Ligands or heteroatom groups that are not part of any polymer (protein or nucleic acid) in the structure may also have missing atoms or atoms with zero occupancy. In such instances the name of the heteroatom group or ligand, chain ID and model number (if applicable) will be listed in _pdbx_unobs_or_zero_occ_atoms with polymer_flag (N) and occupancy_flag (1) or if there are atoms with 0.00 occupancy in pdbx_unobs_or_zero_occ_atoms with polymer_flag (N) and occupancy_flag (0).
6. Chain ID assignment
Definition of chain ID
The chain ID is a unique identifier for each macromolecular polymer and all chemical groups (including waters) associated with it. The length accepted is up to four characters.
Which moieties are assigned chain IDs?
All atoms in the coordinate section of the PDB entry will be assigned a chain ID (_atom_site.auth_asym_id).
How will chain IDs be assigned?
Each polymer (polypeptides, polynucleotides, or oligosaccharides) is assigned a unique chain ID, usually the chain IDs of polypeptides and polynucleotides are provided by authors while the chain IDs of oligosaccharides are assigned by the wwPDB. Chain IDs for all bound moieties and waters are assigned based on their proximity (number of contacts) to the nearest polypeptides and polynucleotides. For example, all waters and chemical groups around a protein/DNA/RNA polymer are assigned the chain ID of the polymer they surround. If a polymer chain was cleaved before or during the experiment, each fragment of the polymer should be assigned a unique chain ID.
Why are chain IDs assigned in this way?
The wwPDB has established this rule to improve the usability and interpretation of the structural data. Assigning the same chain ID for all moieties associated with a polymer enables rapid and uniform identification of feature analysis.
How are chain IDs assigned to chemical groups and waters?
All chemical groups and waters will be assigned the same chain ID as the nearest polypeptide or polynucleotide chain. If a particular chemical group/water is equidistant from more than one chain, then the chain ID is randomly assigned to be that of any one of these polymers. Waters further than 5 Angstroms away from the polymer that can be moved by symmetry to within 5 Angstroms, will be automatically moved during entry processing. Waters further than 5 Angstroms away from any polymer, which cannot be brought closer to a polymer chain by application of symmetry, will be brought to the attention of the depositor. These waters will be listed in _pdbx_distant_solvent_atoms cif category with the distance to the nearest macromolecule.
loop_
_pdbx_distant_solvent_atoms.id
_pdbx_distant_solvent_atoms.PDB_model_num
_pdbx_distant_solvent_atoms.auth_atom_id
_pdbx_distant_solvent_atoms.label_alt_id
_pdbx_distant_solvent_atoms.auth_asym_id
_pdbx_distant_solvent_atoms.auth_comp_id
_pdbx_distant_solvent_atoms.auth_seq_id
_pdbx_distant_solvent_atoms.PDB_ins_code
_pdbx_distant_solvent_atoms.neighbor_macromolecule_distance
_pdbx_distant_solvent_atoms.neighbor_ligand_distance
1 1 O ? A HOH 1146 ? 6.79 .
2 1 O ? B HOH 1113 ? 5.96 .
How are chain IDs related to residue numbering?
All residues and chemical groups in a file should be uniquely identified. Once the polymers and chemical groups associated with them are assigned chain IDs, the numbering of all residues, chemical groups and waters for each chain ID must be unique. Numbering of residues that were not modelled due to limited experimental data should also be considered here. The wwPDB encourages deposition of polymer chains with sequential residue numbering. For protein chains, the authors are encouraged to follow the UniProt residue numbering, wherever possible. The use of non-sequential residue numbering and insertion codes should be avoided as much as possible in order to make structures easily interpretable by the larger scientific community. If the coordinate residue numbers, as provided by the author, are unique and sequential within a particular chain ID, the residues will not be renumbered. If the author has already correctly sorted ligands to polymer chains with which they are associated, these are not reassigned.
How polymer chain IDs should be assigned?
Upper- and lower-case letters, numbers (0-9) and a combination of these should be used for chain IDs. It is preferred that lower case letters and combination of letters/numbers are used after all upper letters and numbers have been used. Symbols should never be used for chain IDs.
Consistent use of chain IDs in PDB structures and manuscripts
During the annotation of structures, the numbering and chain IDs of the chemical groups and waters associated with the polymeric chains may be changed in accordance with the wwPDB chain ID rules. The wwPDB strongly encourages depositors to use the wwPDB-assigned chain ID and residue numbers in any publication material. Deposition and processing of structures prior to preparation of the manuscript will ensure consistent usage of chain IDs and residue numbers in the manuscript and the PDB file. Validation and structure analysis reports generated during annotation may also be helpful in the manuscript preparation.
Structures not compatible with PDB format
Structures that cannot be represented in the legacy PDB file format (PDB incompatible), for example, structures containing multiple character chain ids, >62 chains, PDB incompatible secondary structure records and/or >99999 ATOM lines, will be available in the PDB archive as single PDBx/mmCIF files that represent the entire structure. In addition, these structures will have TAR files containing a collection of best effort/minimal files in the PDB file format to support users and software tools that rely solely on the PDB file format. The _pdbx_database_status.pdb_format_compatible flag will be set to N in the mmCIF files for such structures:
_pdbx_database_status.pdb_format_compatible
7. HEADER assignment
How is the header keyword (_struct_keywds.pdbx_keywords) record assigned?
One keyword is used to briefly describe the broad function of the macromolecules present in the PDB entry. A list of classifications is available at the end of this document and at https://files.wwpdb.org/pub/pdb/doc/format_descriptions/class.dat.
Header functions are assigned by the annotator
The annotator typically assigns the function after reviewing the UniProt keywords and keywords provided by the author. The annotated header will also be included in the entry keywords (_struct_keywords.text) and if the header is a complex (/ separated), the word ‘complex’ will be added in the keywords. If protein is an enzyme, the general class of enzymes is used. For example, the header is assigned as oxidoreductase if the E.C. number starts with 1. See Appendix A or https://files.wwpdb.org/pub/pdb/doc/format_descriptions/class.dat for the list of available headers.
- If protein has no known function, "UNKNOWN FUNCTION" is used.
- If the function is putative, such as a "putative hydrolase" the header will be assigned based on the putative assignment, i.e., "HYDROLASE".
- If the annotator is unsure of the function, the annotator asks the author to choose the appropriate header from within the standard header list.
- If the function is new to the PDB, a generalized header describing the function will be added to the standard header list.
Two or more HEADERs can be combined using the following rules:
- For macromolecular complexes, the headers are separated with a forward slash (HEADER1/HEADER2). e.g., TRANSCRIPTION/DNA (Note that protein is listed first in protein/nucleic acid complexes).
- The word "complex" will not be used in the header record but will appear in the keywords record whenever there is a "/" in the header.
- A multifunctional macromolecule will have commas between the different headers: HEADER1,HEADER2
- Functions such as inhibitor, activator, receptor, substrate of a macromolecule can be added to existing headers (HEADER INHIBITOR) e.g., HYDROLASE INHIBITOR, HYDROLASE ACTIVATOR, HYDROLASE RECEPTOR, HYDROLASE REGULATOR.
How are keywords (_struct_keywords.text) assigned?
Keywords are provided by the entry author. The header keyword (from _struct_keywds.pdbx_keywords) record will also be listed here.
8. Assembly
Quaternary assembly (_pdbx_struct_assembly, _pdbx_struct_assembly_gen, _pdbx_struct_oper_list)
The quaternary structure in a PDB entry may include software calculated quaternary assembly and/or author determined biological assembly, or the biologically relevant form of the molecule for which there is experimental evidence.
We derive a likely oligomeric state of the structure based on the surface area of interactions and the association of macromolecules. Frequently, this derived structure and the biological assembly are the same. However, due to crystal packing forces, experimental evidence, or author opinion, the biological assembly and the derived oligomeric structure may be different. For example, a macromolecule may be assigned a hexameric quaternary structure in the crystal, but the biological assembly may be monomeric. It is recognized that quaternary structure programs such as PISA1 and PQS2 do not work with every case, such as antibodies, or any case where a biological process has a low association/dissociation property. The quaternary assembly is calculated and evaluated by the annotation staff, in addition to the biological assembly information provided by the author, known from the literature or similar structures in the PDB archive.
The matrices forming the quaternary structure will be reported in _pdbx_struct_oper_list mmCIF category and will be assigned by the wwPDB annotators. In instances where the author's description of the biological assembly disagrees with what the crystal structure appears to present, the biological assembly can be chosen by the depositor, and reported in the file. The total surface area, buried surface area and free energy gain will be listed if two polymers have an interface.
Nomenclature: The number of subunits in an oligomeric complex are described using names that end in -mer (Greek for "part, subunit"). Formal Greco-Latinate names are generally used for the first ten types and can be used for up to twenty subunits, whereas higher order complexes are usually described by the number of subunits, followed by -meric.
In pdbx_struct_assembly.oligomeric_details and pdbx_struct_assembly.oligomeric_count, any polypeptide of length of 3 or more amino acids or 2 or more nucleotides is considered in the naming of a quaternary structure as monomeric or dimeric etc.:
| 1 = monomeric |
8 = octameric |
15 = pentadecameric |
| 2 = dimeric |
9 = nonameric |
16 = hexadecameric |
| 3 = trimeric |
10 = decameric |
17 = heptadecameric |
| 4 = tetrameric |
11 = undecameric |
18 = octadecameric |
| 5 = pentameric |
12 = dodecameric |
19 = nonadecameric |
| 6 = hexameric |
13 = tridecameric |
20 = eicosameric |
| 7 = heptameric |
14 = tetradecameric |
21-meric etc. |
Please note that the multi-mer described in PDB remark 350 represents either homo or hetero multi-mer for that entry.
9. Miscellaneous records
File versioning and revision history
The wwPDB maintains a file versioning system that allows Depositors of Record to update their own previously released entries.
Version numbers of each PDB archive entry is designated using a #-# identifier. The first digit specifies the major version, and the second designates the minor version. The Structure of Record (i.e., the initial set of released atomic coordinates) is designated as Version 1-0. Thereafter, the major version digit is incremented with each substantial revision of a given entry (e.g., Version 2-0, when the atomic coordinates are replaced for the first time by the Depositor of Record). “Major version changes” are defined as updates to the atomic coordinates, polymer sequence(s), and/or chemical identify of a ligand. All other changes are defined as “minor changes”. When a major change is made, the minor version number is reset to 0 (e.g., 1-0 to 1-1 to 2-0). The wwPDB retains all major versions with the latest minor versions of an entry within the PDB archive.
Special audit categories are used to capture details of changes to files down to the category level for entry revisions:
http://mmcif.wwpdb.org/dictionaries/mmcif_pdbx_v50.dic/Groups/audit_group.html
For example:
loop_
_pdbx_audit_revision_history.ordinal
_pdbx_audit_revision_history.data_content_type
_pdbx_audit_revision_history.major_revision
_pdbx_audit_revision_history.minor_revision
_pdbx_audit_revision_history.revision_date
1 'Structure model' 1 0 2020-03-25
2 'Structure model' 1 1 2020-04-08
3 'Structure model' 1 2 2020-05-06
#
_pdbx_audit_revision_details.ordinal 1
_pdbx_audit_revision_details.revision_ordinal 1
_pdbx_audit_revision_details.data_content_type 'Structure model'
_pdbx_audit_revision_details.provider repository
_pdbx_audit_revision_details.type 'Initial release'
_pdbx_audit_revision_details.description ?
_pdbx_audit_revision_details.details ?
#
loop_
_pdbx_audit_revision_group.ordinal
_pdbx_audit_revision_group.revision_ordinal
_pdbx_audit_revision_group.data_content_type
_pdbx_audit_revision_group.group
1 2 'Structure model' 'Database references'
2 2 'Structure model' 'Structure summary'
3 3 'Structure model' 'Database references'
4 3 'Structure model' 'Source and taxonomy'
5 3 'Structure model' 'Structure summary'
#
loop_
_pdbx_audit_revision_category.ordinal
_pdbx_audit_revision_category.revision_ordinal
_pdbx_audit_revision_category.data_content_type
_pdbx_audit_revision_category.category
1 2 'Structure model' entity
2 2 'Structure model' pdbx_related_exp_data_set
3 3 'Structure model' entity
4 3 'Structure model' entity_name_com
5 3 'Structure model' entity_src_gen
6 3 'Structure model' struct_ref
7 3 'Structure model' struct_ref_seq
#
loop_
_pdbx_audit_revision_item.ordinal
_pdbx_audit_revision_item.revision_ordinal
_pdbx_audit_revision_item.data_content_type
_pdbx_audit_revision_item.item
1 2 'Structure model' '_entity.pdbx_description'
2 3 'Structure model' '_entity.pdbx_description'
3 3 'Structure model' '_entity.pdbx_ec'
4 3 'Structure model' '_entity_src_gen.gene_src_common_name'
5 3 'Structure model' '_entity_src_gen.pdbx_gene_src_gene'
6 3 'Structure model' '_struct_ref.db_code'
7 3 'Structure model' '_struct_ref.db_name'
8 3 'Structure model' '_struct_ref.pdbx_align_begin'
9 3 'Structure model' '_struct_ref.pdbx_db_accession'
10 3 'Structure model' '_struct_ref.pdbx_seq_one_letter_code'
11 3 'Structure model' '_struct_ref_seq.db_align_beg'
12 3 'Structure model' '_struct_ref_seq.db_align_end'
13 3 'Structure model' '_struct_ref_seq.pdbx_db_accession'
Link records (_struct_conn, LINK, SSBOND)
LINK records will be automatically generated using standard software. There are various cutoff distances specified in this software for various kinds of LINK records. LINK records can include covalent bonding, metal coordination etc. LINK records may also be added by the author.
SSBOND records are created for cysteine residues involved in disulfide bonds and do not include disulfide bonds between other residues or ligands.
Standard software will automatically add the struct_conn category to the file, listing the distances of the link records between metal ions and surrounding residues following standard coordination geometry. Below is an excerpt of LINK records in PDB entry 1W3M.
mmCIF format:
loop_
_struct_conn.id
_struct_conn.conn_type_id
_struct_conn.pdbx_leaving_atom_flag
_struct_conn.pdbx_PDB_id
_struct_conn.ptnr1_label_asym_id
_struct_conn.ptnr1_label_comp_id
_struct_conn.ptnr1_label_seq_id
_struct_conn.ptnr1_label_atom_id
_struct_conn.pdbx_ptnr1_label_alt_id
_struct_conn.pdbx_ptnr1_PDB_ins_code
_struct_conn.pdbx_ptnr1_standard_comp_id
_struct_conn.ptnr1_symmetry
_struct_conn.ptnr2_label_asym_id
_struct_conn.ptnr2_label_comp_id
_struct_conn.ptnr2_label_seq_id
_struct_conn.ptnr2_label_atom_id
_struct_conn.pdbx_ptnr2_label_alt_id
_struct_conn.pdbx_ptnr2_PDB_ins_code
_struct_conn.ptnr1_auth_asym_id
_struct_conn.ptnr1_auth_comp_id
_struct_conn.ptnr1_auth_seq_id
_struct_conn.ptnr2_auth_asym_id
_struct_conn.ptnr2_auth_comp_id
_struct_conn.ptnr2_auth_seq_id
_struct_conn.ptnr2_symmetry
_struct_conn.pdbx_ptnr3_label_atom_id
_struct_conn.pdbx_ptnr3_label_seq_id
_struct_conn.pdbx_ptnr3_label_comp_id
_struct_conn.pdbx_ptnr3_label_asym_id
_struct_conn.pdbx_ptnr3_label_alt_id
_struct_conn.pdbx_ptnr3_PDB_ins_code
_struct_conn.details
_struct_conn.pdbx_dist_value
_struct_conn.pdbx_value_order
covale1 covale both ? M LNG . C1 A ? ? 1_555 A ASP 1 N ? ? A LNG 0 A ASP 1 1_555 ? ? ? ? ? ? ? 1.306 ?
covale2 covale both ? M LNG . C1 B ? ? 1_555 A ASP 1 N ? ? A LNG 0 A ASP 1 1_555 ? ? ? ? ? ? ? 1.350 ?
covale3 covale one ? A ASP 1 C ? ? ? 1_555 A VLL 2 N ? ? A ASP 1 A VLL 2 1_555 ? ? ? ? ? ? ? 1.334 ?
covale4 covale both ? A VLL 2 C ? ? ? 1_555 A CPI 3 N ? ? A VLL 2 A CPI 3 1_555 ? ? ? ? ? ? ? 1.340 ?
metalc1 metalc ? ? A VLL 2 O ? ? ? 1_555 N CA . CA ? ? A VLL 2 A CA 3013 1_555 ? ? ? ? ? ? ? 2.335 ?
covale5 covale one ? A VLL 2 NG2 ? ? ? 1_555 A PRO 11 C ? ? A VLL 2
Secondary structure records
Helix and sheet records are automatically generated by the Promotif algorithm, and stored in the struct_conf, struct_sheet, struct_sheet_order, struct_sheet_range and pdbx_struct_sheet_hbond categories. Authors who wish to provide their own helix and sheet records may do so.
From June 2021, PDB formatted files are no longer generated for PDB entries where the sheet topology cannot be generated in the PDB format (complex beta sheet topologies where the definition of the strands within a beta sheet cannot be presented in a linear description). This limitation, however, is not an issue in the PDBx/mmCIF formatted file, where these complex beta sheet topologies can be correctly captured. Therefore, for these examples, wwPDB will continue to provide secondary structure with helix and sheet information in the PDBx/mmCIF formatted file.
Validation, calculation of bond, angle, torsion deviations, etc.
The calculation of bond and angle deviations for protein entries will be based on the updated Engh & Huber amino acid target values3. For nucleic acids, the Parkinson et al., statistics are
used for these calculations4. All bonds and angles that deviate more than 6 times from their standard target values will be flagged as a deviation. The PHI/PSI values are based on Kleywegt’s calculations5.
The deviations are reported in mmcif categories below, and REMARK 500 of PDB format file.
CLOSE CONTACTS IN SAME ASYMMETRIC UNIT (_pdbx_validate_close_contact)
SYMMETRY RELATED CLOSE CONTACTS (_pdbx_validate_symm_contact)
BOND LENGTHS (_pdbx_validate_rmsd_bond)
BOND ANGLES (_pdbx_validate_rmsd_angle)
TORSION ANGLES (_pdbx_validate_torsion)
NON-CIS, NON-TRANS (_pdbx_validate_peptide_omega)
SIDE CHAIN PLANAR GROUPS (_pdbx_validate_planes)
MAIN CHAIN PLANARITY (protein only) (_pdbx_validate_main_chain_plane)
CHIRAL CENTERS (protein C-alpha only) (_pdbx_validate_chiral)
Additional entry details
Additional entry details are annotated in _pdbx_entry_details, such as additional functional details of the compounds in _pdbx_entry_details.compound_details (REMARK 400), additional polymer sequence details in _pdbx_entry_details.sequence_details (REMARK 999), and any other information about the ligand in _pdbx_entry_details.nonpolymer_details (REMARK 600).
Related entries
Authors may provide information about related entries (pdbx_database_related, REMARK 900) to relate other entries to the current entry.
Deprecation of _struct_site (SITE) records
In June 2021 the in-house legacy software which produced _struct_site and _struct_site_gen records was retired. At that point, the wwPDB no longer generated these categories for newly deposited PDB entries, however existing entries were unaffected.
10. Structural Genomics Entries
Structural genomics (SG) entries are usually either X-ray or NMR structures deposited by the various structural genomics groups around the world. In the USA, these structures are deposited primarily by the several Protein Structure Initiative (PSI) groups. Europe, UK, Canada and Japan also have several structural genomics centers. The SG structures are processed in the same way as any other PDB deposition. There are just a few additional rules for the annotation of these entries. These are listed below:
- Usually, the SG entries are deposited with a "release immediately" status. In special instances (like for the CASP competition) the depositions may be processed and held for a pre-determined period before its release.
- For entries deposited by an SG group, the author list also includes the name of the SG center (like JCSG, MCSG, BSGC, etc.).
- The following words and phrases are also included in the keywords: SG center name (in full and also the abbreviation), Structural Genomics. If the entry is from a PSI center, the initials "PSI" and the words "Protein Structure Initiative" are added to the
keywords. Entries that are part of the second phase of the PSI project are labelled PSI-2.
- For SG entries deposited by centers which also deposit targets to TargetDB, the TargetDB ID for each sequence in the entry is included in the file and it appears in _pdbx_database_related (REMARK 900).
- The project name, center name and center abbreviation are included in the _pdbx_SG_project mmCIF category.
11. Information specific to X-ray structures
Deposition of X-ray structures
All structures determined by crystal X-ray diffraction where the structure is that of a non-virus capsid should contain the atomic coordinates for the whole crystallographic asymmetric unit (ASU). The ASU is defined as the smallest unit that can be rotated and translated to generate one unit cell using only the symmetry operators allowed by the crystallographic symmetry. The asymmetric unit may be one molecule or one subunit of a multimeric protein, but it can also be more than one.
Information contained in structure factor files
Structure factor (sf) files should include information such as cell, space group, symmetry, wavelength, and number of reflections of the entry. The date included in the header of the sf file will be the date of release, not the deposition date. If authors include multiple structure factor files (such as a set for refinement, multiple sets for phasing, etc.), the data will be archived as multiple data blocks. For example, the reflections for refinement will be listed in the category '_refln' and the phasing data sets will be listed in the category '_phasing_set_refln.' The different data sets can be distinguished by crystal and/or wavelength ID as appropriate. If different cell dimensions are present, this information will also be included in the file. The dataset used for the refinement should be listed as a first data block and should contain diffraction indices h,k,l, observed amplitudes and/or intensities, their respective sigma values and refinement test set.
See Appendix B for examples of the structure factor files.
Refinement and data collection statistics significant figures
The values provided by the author will be retained in the mmCIF and PDB files.
Hydrogens in crystal structures
Hydrogens in crystal structures will be retained regardless of resolution with the occupancy provided by the depositor.
Matthews coefficient and solvent content
For crystal structures, the Matthews coefficient and solvent content will be automatically calculated using the following equations:
Matthews coefficient6 = volume of unit cell/(the molecular weight of macromolecule*Y*X) Where Y is the number of asymmetric units in the unit cell (i.e., the number of symmetry operators in the space group). The unknown variable, X, is the number of molecules in the asymmetric unit.
Solvent content = 1 - 1.23/ (Matthews coefficient)
The molecular weight includes protein and nucleic acids based on sequences, no water and ligands. In cases of viral capsids and proteolytic fragments, the Matthews coefficient and solvent content should be author provided and will not be automatically calculated.
Number of reflections
The number of reflections for refinement is the number of crystallographically unique measured reflections that satisfy both the resolution and the observation limits. The number of reflections for data collection is the total number of crystallographically unique measured reflections that are labelled as observed by the criterion on sigma(I) or on sigma(F). The number of reflections reported for refinement should be less than or equal to the number reported for data collection, even if Friedel pairs were used.
Twinned structures
Structures based on twinned crystal diffraction data can be identified through use of the twinning tokens to the mmCIF public exchange dictionary, pdbx_twin. The tokens can be used for identification of twinning operator, type, and fraction.
The structure factor file for a twinned structure should include the twinned data used for refinement first. If the authors have the detwinned data, it may also be included in the file. The pdbx_reflns_twin tokens should be included in the structure factor file.
MAD data
If MAD data were collected, authors are encouraged to provide all data sets used in structure solution and refinement. The data set used for refinement should be listed first in the structure factor file. Authors are encouraged to provide the other (phasing) datasets. Wavelengths, source and other data collection information for all data sets should also be provided.
BioSync and information about synchrotron data collection
Information about synchrotron sources and beamlines will be made consistent with the standard names used in the BioSync database (http://biosync.rcsb.org/).
12. Information specific to NMR structures
Pseudoatoms (Q atoms)
Pseudoatoms (also known as Q atoms) submitted for NMR entries will be removed from the entry.
Superimposed models
At least one domain of the NMR entry should be superimposed across all models. It is recognized that for multi-domain NMR structures, domain movements prevent the whole structure from being aligned through the length of the molecule. However, in order to highlight the relative movement of the domains, it makes sense to superpose at least one part of the structure across all the models deposited under a PDB accession code. This does not detract from the scientific value of the coordinate set or the experiment, but on the contrary, serves to highlight domain motion with respect to a fixed point. The superposition need not be arbitrary but may be done at the choosing of the depositor. This will allow the larger scientific community easy identification of the protein folds. It also facilitates identification of model variations across different parts of the structure.
Experimental Methods: There are two types of NMR experimental methods (EXPDTA, _exptl.method):
SOLID-STATE NMR SOLUTION NMR
SOLUTION NMR
Homogeneity of Ensemble
All models in a deposition should be superimposed in an appropriate author determined manner and only one superposition method should be used. Structures from different experiments, or different domains of a structure, should not be superimposed and deposited as models of a deposition.
All models in an NMR ensemble must be homogeneous - each model must have the exact same atoms (hydrogen and heavy atoms), sequence and chemistry.
Deposition of minimized average structure must be accompanied with ensemble and must be homogeneous with ensemble.
Model type
MDLTYP record contains additional annotation pertinent to the coordinates presented in the entry. This record will indicate minimized average structure with model number of the minimized average structure. The corresponding cif is _struct.pdbx_model_type_details. MDLTYP MINIMIZED AVERAGE, MODEL X
Best representative conformer
This record defines the best representative conformer in the ensemble (pdbx_nmr_representative.conformer_id, REMARK 210).
Example:
_pdbx_nmr_representative.conformer_id 1
_pdbx_nmr_representative.selection_criteria "lowest energy"
During the annotation of structures, the best representative conformer is moved to MODEL 1 since HELIX/SHEET, SITE and LINK records are generated based on the first model in the coordinates. Authors are notified of how the annotation staff handled this case.
13. Information specific to Electron Microscopy structures
There are two types of general experimental methods for 3DEM: electron microscopy (EM; comprises single particle and helical reconstruction techniques, tomography, and subtomogram averaging) and electron crystallography (EC). There are three main data file types associated with 3DEM:
- 3D volume, or map
- Atomic coordinates
- Structure factors (EC only)
An EM deposition may comprise map only, coordinates and map, or only coordinates. Any atomic coordinates deposited must have an associated map, i.e., the map from which those coordinates were derived. That associated map must be the either the primary (see Map Information section below) map of the shared deposition (in the case of a deposition containing both map and coordinates) or the primary map of a different deposition. In any case, the EMDB ID of the map must be included in _pdbx_database_related (REMARK 900) as “associated EM volume” (any other related maps will be designated as “other EM volume”). In addition, no EM atomic coordinates will be released to the PDB without simultaneous or previous release of its associated map to the EMDB.
An EC deposition will generally comprise only coordinates and structure factors, analogous to X-ray crystallography. Occasionally, a map will also be included in the deposition or associated from a different deposition. In the absence of structure factors, there must be an associated map. Maps for EC depositions must be treated identically to maps for EM depositions.
In addition to the three main data file types, there are some auxiliary file types:
- Map image file (mandatory for depositions that include maps)
- FSC curve XML (optional; should be the FSC curve used to estimate the reported resolution)
- Layer lines (optional for tomography entries)
Map Information
Maps are accepted in two formats, CCP4 and MRC, and will be converted to CCP4 format upon deposition. There are three depositor-provided data items associated with each map: the voxel size (in pixels / Angstrom; mandatory for all maps), the recommended contour level (for optimal display of isocontour maps; mandatory for all maps except tomograms), and annotation details (a brief description of the map; optional for all maps). There are four different subtypes assigned to maps:
- Primary map.The primary map is the center of a map deposition (and EMDB entry). If maps are deposited, a primary map is mandatory. Each deposition will have only one primary map, and map validation presented by the wwPDB will be focused on the primary map. If deposited coordinates are derived from a map, that map must be a primary map.
- Half map. Half maps are the two maps (based on alternate halves of the data set) used for cross-validation during reconstruction of the primary map. Deposition of half maps is optional but encouraged. If half maps are provided, there must be two and only two, and they should be unaltered and uncropped.
- Mask. Masks are used during map processing for removing noise, improving map quality, isolating portions of maps (segmenting), etc. Deposition of masks is optional, and any number of masks can be included as part of a deposition.
- Additional map. Any maps included in a deposition that are not the primary map, the half maps associated with the primary map, or masks are labeled “additional maps”. These can include raw maps, unmasked maps, segmentations, etc. Deposition of additional maps is optional, and any number of additional maps can be included as part of a deposition.
EM Metadata
Additional metadata are collected for 3DEM depositions relative to other PDB depositions. Generally, these involve the equipment and parameters related to 3DEM data collection and 3D volume reconstruction, as well as refinement of atomic coordinates where relevant. In PDBx/mmCIF, these metadata are designated _em_* (where * is a wildcard). They correspond to metadata collected for use by EMDB, though some but not all of the metadata will be also be used by the PDB.
14. Viral capsids and other complex assemblies
For the purposes of annotation, a complex assembly is defined as a structure for which the full biological assembly and/or crystallographic asymmetric unit is built by applying a set of non-crystallographic rotation/translation transformations to a set of deposited coordinates.
Icosahedral Viruses
The icosahedral virus is the most common complex assembly deposited to the PDB. The author generally deposits the coordinates of the icosahedral asymmetric unit and supplies a set of 60 transformation matrices to be applied to the coordinates to produce the full biological assembly. We will continue to request these matrices from the authors. From the author-provided matrices and coordinates we will calculate a standard set of 60 ordered matrices as well as the transformation that moves the complex to the standard icosahedral frame (same frame used by ViperDB). The calculated matrices, the frame transformation, and the description of how they are to be applied to the coordinates to build the assembly will be stored in _pdbx_struct records.
For crystal structures we will also request a complete description of how to build the crystal asymmetric unit, and the description will be archived in _pdbx_struct records. If the coordinates are provided in the crystal frame, the non-crystallographic symmetry transformations will also be placed in struct_ncs_oper records and will appear in MTRIX records, enabling validation against the structure factor data.
Regular Symmetries
Icosahedral point symmetry is just one type of symmetry that can be adopted by a complex assembly. Other point symmetries (see table below) or helical symmetries are possible. For all structures deposited as complex assemblies, we will archive symmetry information as appropriate in _pdbx_point_symmetry or _pdbx_helical_symmetry records.
| point symmetry |
Schoenflies symbol |
# equivalent positions |
| circular |
Cn |
n |
| dihedral |
Dn |
2n |
| tetrahedral |
T |
12 |
| octahedral |
O |
24 |
| icosahedral |
I |
60 |
From: International Tables for Crystallography, Volume A, 4th edition, Table 10.4.2, p. 782-783
Point symmetry
The point symmetry information is stored in the pdbx_point_symmetry category (REMARK 300).
Example:
_pdbx_point_symmetry.entry_id 2BK1
_pdbx_point_symmetry.Schoenflies_symbol C
_pdbx_point_symmetry.circular_symmetry 38
T = TETRAHEDRAL
D = DIHEDRAL
O = OCTAHEDRAL
I = ICOSAHEDRAL
15. Re-refinement of another author's data
The following text is for a dedicated remark for cases where an author re-refines another author's data (pdbx_database_remark, REMARK 0). The remark will always appear in entries where the author refined someone else's data. The entry will be treated as an experimental structure.
Example of the entry 1ZET, based on the data of 1T3N:
_pdbx_database_remark.id 0
_pdbx_database_remark.text
;THIS ENTRY 1ZET REFLECTS AN ALTERNATIVE MODELING OF THESTRUCTURAL DATA IN R1T3NSF ORIGINAL DATA DETERMINED BYAUTHOR: D.T.NAIR,R.E.JOHNSON,S.PRAKASH,L.PRAKASH,A.K.AGGARWAL
;
#
Original author's paper in will be listed in citation category with citation_id = original_data_1:
loop_
_citation.id
_citation.title
_citation.journal_abbrev
_citation.journal_volume
_citation.page_first
_citation.page_last
_citation.year
_citation.journal_id_ASTM
_citation.country
_citation.journal_id_ISSN
_citation.journal_id_CSD
_citation.book_publisher
_citation.pdbx_database_id_PubMed
_citation.pdbx_database_id_DOI
primary 'DNA polymerases: Hoogsteen base-pairing in DNA replication?' Nature 437 E6 '7; discussion E7' 2005 NATUAS UK 0028-0836 0006 ? 16163299 10.1038/nature04199
original_data_1 'Replication by human DNA polymerase-iota occurs by Hoogsteen base-pairing.' Nature 430 377 380 2004 NATUAS UK 0028-0836 0006 ? 15254543 10.1038/nature02692
#
loop_
_citation_author.citation_id
_citation_author.name
_citation_author.ordinal
primary 'Wang, J.' 1
original_data_1 'Nair, D.T.' 2
original_data_1 'Johnson, R.E.' 3
original_data_1 'Prakash, S.' 4
original_data_1 'Prakash, L.' 5
original_data_1 'Aggarwal, A.K.' 6
Note: In entries where pdbx_database_remark (REMARK 0) is included as described above, pdbx_database_related (REMARK 900) will also reflect the reuse of existing experimental data as shown in the example below:
#
_pdbx_database_related.db_name PDB
_pdbx_database_related.db_id 1T3N
_pdbx_database_related.details 'Structure of the catalytic core of DNA polymerase Iota in complex with DNA and dTTP'
_pdbx_database_related.content_type re-refinement
The SF file has the following, with additional information on what the author added, if applicable:
_audit.revision_id 1_0
_audit.creation_date 2005-07-19
_audit.update_record
;Initial release, author used sf file from pdb entry 1t3n, and added columns Fcalc, phases and FOM
;
Appendices:
A. HEADER list (struct_keywords.pdbx_keywords)
Below is the list of headers to be used when processing PDB entries:
ALLERGEN
ANTIBIOTIC (peptidic, saccharide containing)
ANTIFREEZE PROTEIN
ANTIFUNGAL PROTEIN
ANTIMICROBIAL PROTEIN
ANTITOXIN
ANTITUMOR PROTEIN
ANTIVIRAL PROTEIN
APOPTOSIS
ATTRACTANT
BIOSYNTHETIC PROTEIN
BLOOD CLOTTING
CARBOHYDRATE
CELL ADHESION
CELL CYCLE
CELL INVASION
CHAPERONE
CIRCADIAN CLOCK PROTEIN
CONTRACTILE PROTEIN
CYTOKINE (includes interleukins, interferons)
DE NOVO PROTEIN (ARTIFICIALLY DESIGNED, OFTEN SYNTHETIC)
DNA
DNA-RNA HYBRID (used when biological assembly contains mixed DNA and RNA residues or strands)
ELECTRON TRANSPORT
ENDOCYTOSIS
EXOCYTOSIS
FLAVOPROTEIN
FLUORESCENT PROTEIN
GENE REGULATION (use only when TRANSCRIPTION, REPLICATION,
TRANSLATION are not applicable)
HORMONE
HYDROLASE (E.C.3.-.-.-)
IMMUNE SYSTEM (includes antibodies, antigens)
IMMUNOSUPPRESSANT
ISOMERASE (E.C.5.-.-.-)
LIGASE (E.C.6.-.-.-)
LIPID TRANSPORT
LUMINESCENT PROTEIN
LYASE (E.C.4.-.-.-)
MEMBRANE PROTEIN (no other function known)
METAL TRANSPORT
MOTOR PROTEIN
NEUROPEPTIDE
ONCOPROTEIN
OXIDOREDUCTASE (E.C.1.-.-.-)
OXYGEN BINDING
OXYGEN STORAGE
OXYGEN TRANSPORT
PHOTOSYNTHESIS
PLANT PROTEIN (no other function known)
PROTON TRANSPORT
PROTEIN TRANSPORT (a protein involved in transporting other protein)
RECOMBINATION
REPLICATION
RIBOSOME (use only when TRANSLATION is not correct; do not specify /RNA even when present!)
RIBOSOMAL PROTEIN
RNA
SIGNALING PROTEIN (includes G-proteins)
SPLICING
STRUCTURAL GENOMICS (product of a probable gene)
STRUCTURAL PROTEIN
SURFACTANT PROTEIN
TOXIN (not antibiotic, can use e.g. HYDROLASE INHIBITOR, TOXIN)
TRANSFERASE (E.C.2.-.-.-)
TRANSCRIPTION (DNA to RNA)
TRANSLATION (protein synthesis; prefer over RIBOSOME)
TRANSLOCASE (E.C.7.-.-.-)
TRANSPORT PROTEIN (a protein that transports anything)
NUCLEAR PROTEIN (whether involved in binding RNA/DNA or some sort of nuclear processing is unclear)
VIRUS (for entire viral capsid)
VIRAL PROTEIN (viral protein not involved in the viral capsid)
VIRUS LIKE PARTICLE ((for cases where virus like particles are assembled , but are not the standard virus)
When no other function is known use the following:
CHOLINE-BINDING PROTEIN
CYTOSOLIC PROTEIN (a protein whose function is not known well but is known to be found in the cytosol of a cell.)
DNA BINDING PROTEIN
RNA BINDING PROTEIN
LIPID BINDING PROTEIN
METAL BINDING PROTEIN (such as ZN, FE)
PEPTIDE BINDING PROTEIN
PROTEIN BINDING (implies binding of protein by protein)
SUGAR BINDING PROTEIN xxx-BINDING PROTEIN (for any xxx ligand if none of above applies, such as HEME, AVIDIN, BIOTIN)
PROTEIN FIBRIL
UNKNOWN FUNCTION
B. Format for Structure Factors
Example 1 Structure factor file containing single data set used for refinement
data_rxxxxsf
#
_audit.revision_id 1_0
_audit.creation_date ?
_audit.update_record 'Initial release'
#
#
_entry.id xxxx
#
#
_cell.entry_id xxxx
_cell.length_a 118.8600
_cell.length_b 155.0300
_cell.length_c 155.5400
_cell.angle_alpha 90.0000
_cell.angle_beta 90.0000
_cell.angle_gamma 90.0000
#
_symmetry.entry_id xxxx
_symmetry.Int_Tables_number 20
_symmetry.space_group_name_H-M 'C 2 2 21'
#
loop_
_symmetry_equiv.id
_symmetry_equiv.pos_as_xyz
1 'X, Y, Z'
2 '-X, -Y, Z+1/2'
3 'X, -Y, -Z'
4 '-X, Y, -Z+1/2'
5 'X+1/2, Y+1/2, Z'
6 '-X+1/2, -Y+1/2, Z+1/2'
7 'X+1/2, -Y+1/2, -Z'
8 '-X+1/2, Y+1/2, -Z+1/2'
#
#
_diffrn.id 1
_diffrn.crystal_id 1
_diffrn.details ?
#
_diffrn_radiation_wavelength.id 1
_diffrn_radiation_wavelength.wavelength 0.98100
#
_exptl_crystal.id 1
#
_reflns_scale.group_code 1
#
loop_
_refln.wavelength_id
_refln.crystal_id
_refln.scale_group_code
_refln.index_h
_refln.index_k
_refln.index_l
_refln.status
_refln.F_meas_au
_refln.F_meas_sigma_au
_refln.F_calc
_refln.phase_calc
_refln.fom
_refln.pdbx_HL_A_iso
_refln.pdbx_HL_B_iso
_refln.pdbx_HL_C_iso
_refln.pdbx_HL_D_iso
1 1 1 0 0 6 o 299.0 6.4 1306.2 0.0 0.32 0.33 0 0.00 0.00
1 1 1 0 0 10 o 726.8 15.0 1756.7 180.0 0.99 2.86 0 0.00 0.00
...
#END OF REFLECTIONS
Example 2 Structure factor file containing data sets for final refinement and phasing
#
_audit.revision_id 1_0
_audit.creation_date ?
_audit.update_record 'Initial release'
#
#This file contains two data sets. The first data set is used for refinement.
#The second data set is used for phasing.
#
_entry.id xxxx
#
#
_cell.entry_id xxxx
_cell.length_a 108.7420
_cell.length_b 61.6790
_cell.length_c 71.6520
_cell.angle_alpha 90.0000
_cell.angle_beta 97.1510
_cell.angle_gamma 90.0000
#
_symmetry.entry_id xxxx
_symmetry.Int_Tables_number 5
_symmetry.space_group_name_H-M 'C 1 2 1'
#
loop_
_symmetry_equiv.id
_symmetry_equiv.pos_as_xyz
1 'X, Y, Z'
2 '-X, Y, -Z'
3 'X+1/2, Y+1/2, Z'
4 '-X+1/2, Y+1/2, -Z'
#
#
_diffrn.id 1
_diffrn.crystal_id 1
_diffrn.details “data used for final refinement”
#
_diffrn_radiation_wavelength.id 1
_diffrn_radiation_wavelength.wavelength 1.0
#
_exptl_crystal.id 1
#
_reflns_scale.group_code 1
#
loop_
_refln.wavelength_id
_refln.crystal_id
_refln.scale_group_code
_refln.index_h
_refln.index_k
_refln.index_l
_refln.status
_refln.F_meas_au
_refln.F_meas_sigma_au
1 1 1 -50 0 1 x ? ?
1 1 1 49 5 1 o 37.7 9.4
1 1 1 50 0 0 x ? ?
...
#END
data_rxxxxAsf
#
#This is second data set for phasing.
#
#
#
_cell.entry_id xxxx
_cell.CCP4_wavelength_id 1
_cell.CCP4_crystal_id 1
_cell.length_a 108.742
_cell.length_b 61.689
_cell.length_c 71.652
_cell.angle_alpha 90.000
_cell.angle_beta 91.151
_cell.angle_gamma 90.000
#
_symmetry.entry_id xxxx
_symmetry.Int_Tables_number 5
_symmetry.space_group_name_H-M 'C 1 2 1'
#
loop_
_symmetry_equiv.id
_symmetry_equiv.pos_as_xyz
1 'X, Y, Z'
2 '-X, Y, -Z'
3 'X+1/2, Y+1/2, Z'
4 '-X+1/2, Y+1/2, -Z'
#
#
_diffrn.id 1
_diffrn.crystal_id 1
_diffrn.details “data used for phasing”
#
_diffrn_radiation_wavelength.id 1
_diffrn_radiation_wavelength.wavelength 0.98100
#
_entry.id xxxx
#
_exptl_crystal.id 1
#
_reflns_scale.group_code 1
#
loop_
_refln.wavelength_id
_refln.crystal_id
_refln.scale_group_code
_refln.index_h
_refln.index_k
_refln.index_l
_refln.status
_refln.F_meas_au
_refln.F_meas_sigma_au
_refln.pdbx_anom_difference
_refln.pdbx_anom_difference_sigma
1 1 1 -50 0 1 x ? ? ? ?
1 1 1 -50 0 2 x ? ? ? ?
...
#END OF REFLECTIONS
Example 3 Structure factor file containing data sets for final refinement and scaled unmerged intensities
data_rxxxxsf
#
loop_
_audit.revision_id
_audit.creation_date
_audit.update_record
1_0 2009-12-08 'Initial release'
1_1 2016-08-10 'unmerged data added'
#
_cell.entry_id xxxx
_cell.length_a 44.0630
_cell.length_b 75.9360
_cell.length_c 85.9440
_cell.angle_alpha 90.0000
_cell.angle_beta 90.0000
_cell.angle_gamma 90.0000
#
_diffrn.id 1
_diffrn.crystal_id 1
_diffrn.ambient_temp ?
_diffrn.crystal_treatment ?
_diffrn.details 'amplitudes used for refinement'
#
_diffrn_radiation_wavelength.id 1
_diffrn_radiation_wavelength.wavelength 1.5418
#
_diffrn_reflns.diffrn_id 1
_diffrn_reflns.pdbx_d_res_high 1.769
_diffrn_reflns.pdbx_d_res_low 19.546
_diffrn_reflns.limit_h_max 24
_diffrn_reflns.limit_h_min 0
_diffrn_reflns.limit_k_max 42
_diffrn_reflns.limit_k_min 0
_diffrn_reflns.limit_l_max 48
_diffrn_reflns.limit_l_min 0
_diffrn_reflns.number 28813
_diffrn_reflns.pdbx_number_obs 28799
#
_entry.id xxxx
#
_exptl_crystal.id 1
#
_reflns_scale.group_code 1
#
_symmetry.entry_id xxxx
_symmetry.space_group_name_H-M 'P 21 21 21'
_symmetry.Int_Tables_number 19
#
loop_
_symmetry_equiv.id
_symmetry_equiv.pos_as_xyz
1 'X, Y, Z'
2 'X+1/2, -Y+1/2, -Z'
3 '-X, Y+1/2, -Z+1/2'
4 '-X+1/2, -Y, Z+1/2'
#
#
loop_
_refln.crystal_id
_refln.wavelength_id
_refln.scale_group_code
_refln.index_h
_refln.index_k
_refln.index_l
_refln.status
_refln.F_meas_au
_refln.F_meas_sigma_au
_refln.intensity_meas
_refln.intensity_sigma
_refln.pdbx_I_plus
_refln.pdbx_I_plus_sigma
_refln.pdbx_I_minus
_refln.pdbx_I_minus_sigma
_refln.pdbx_F_plus
_refln.pdbx_F_plus_sigma
_refln.pdbx_F_minus
_refln.pdbx_F_minus_sigma
_refln.pdbx_anom_difference
_refln.pdbx_anom_difference_sigma
1 1 1 0 0 6 o 1376.410 15.01046 10408.40 226.5000 10408.40 226.5000 10408.40 226.5000 1376.410 15.01046 1376.410 15.01046 0.000000 0.000000
1 1 1 0 0 8 o 217.3632 2.853964 259.2000 6.800000 259.2000 6.800000 259.2000 6.800000 217.3632 2.853964 217.3632 2.853964 0.000000 0.000000
1 1 1 0 0 10 o 106.2069 3.010540 62.00000 3.500000 62.00000 3.500000 62.00000 3.500000 106.2069 3.010540 106.2069 3.010540 0.000000 0.000000
1 1 1 0 0 12 o 429.4464 5.076715 1011.800 23.90000 1011.800 23.90000 1011.800 23.90000 429.4464 5.076715 429.4464 5.076715 0.000000 0.000000
...
#END
data_rxxxxAsf
#
_entry.id xxxx
#
_diffrn.id 1
_diffrn.crystal_id 1
_diffrn.details
;scalepack unmerged intensities
;
#
_exptl_crystal.id 1
#
_diffrn_radiation.diffrn_id 1
_diffrn_radiation.wavelength_id 1
_diffrn_radiation_wavelength.id 1
_diffrn_radiation_wavelength.wavelength 1.5418
#
loop_
_diffrn_refln.diffrn_id
_diffrn_refln.wavelength_id
_diffrn_refln.standard_code
_diffrn_refln.scale_group_code
_diffrn_refln.id
_diffrn_refln.index_h
_diffrn_refln.index_k
_diffrn_refln.index_l
_diffrn_refln.intensity_net
_diffrn_refln.intensity_sigma
1 1 1 1 1 0 0 6 10072.3 319.8
1 1 1 1 2 0 0 -6 10746.7 320.8
1 1 1 1 3 0 0 8 253.1 9.5
1 1 1 1 4 0 0 -8 265.6 9.8
...
# END OF REFLECTIONS
1 E. Krissinel and K. Henrick (2005). Detection of Protein Assemblies in Crystals. In: M.R. Berthold et.al. (Eds.): CompLife 2005, LNBI 3695, pp. 163--174. Springer-Verlag Berlin Heidelberg.
2 Henrick, K., and J. M. Thornton. 1998. PQS: a protein quaternary structure file server. Trends. Biochem. Sci. 23:358-361.
3 Structure quality and target parameters. R. A. Engh and R. Huber. International Tables for Crystallography (2006). Vol. F, ch. 18.3, pp. 382-392.
4 "New Parameters for the Refinement of Nucleic Acid Containing Structures." Gary Parkinson, Jaroslav Vojtechovsky, Lester Clowney, Axel Brunger*, and Helen M. Berman. (1996) Acta Cryst. D 52, 57-64
5 "PHI/PSI- Chology: Ramachandran revisited. " GJ Kleywegt and TA Jones (1996) Structure 4, 1395-1400.
6 See Matthews, B.W. 1968. Solvent content of protein crystals. J. Mol. Biol. 33: 491-497 and
http://www.doe-mbi.ucla.edu/~sawaya/tutorials/Characterize/characterize.html